Геномная языковая модель предсказывает функции белков
МЕНЮ
Главная страница
Поиск
Регистрация на сайте
Помощь проекту
Архив новостей
ТЕМЫ
Новости ИИ
Городские сумасшедшие
ИИ в медицине
ИИ проекты
Искусственные нейросети
Искусственный интеллект
Слежка за людьми
Угроза ИИ
Компьютерные науки
Машинное обуч. (Ошибки)
Машинное обучение
Машинный перевод
Нейронные сети начинающим
Психология ИИ
Реализация ИИ
Реализация нейросетей
Создание беспилотных авто
Трезво про ИИ
Философия ИИ
Генетические алгоритмы
Капсульные нейросети
Основы нейронных сетей
Распознавание лиц
Распознавание образов
Распознавание речи
Творчество ИИ
Техническое зрение
Чат-боты
Авторизация
2024-04-06 11:52

Расшифровка связи между геном и его геномным контекстом имеет фундаментальное значение для понимания и разработки биологических систем. Машинное обучение показало многообещающие результаты в изучении скрытых взаимосвязей, лежащих в основе парадигмы последовательность-структура-функция, на основе массивных наборов данных о последовательностях белков. Однако на сегодняшний день предпринимаются ограниченные попытки расширить этот континуум, включив в него информацию о геномном контексте более высокого порядка. Эволюционные процессы диктуют специфичность геномных контекстов, в которых ген обнаруживается на филогенетических расстояниях, и эти возникающие геномные паттерны можно использовать для выявления функциональных связей между генными продуктами. Здесь мы обучаем модель геномного языка (gLM) на миллионах метагеномных каркасов, чтобы изучить скрытые функциональные и регуляторные отношения между генами. gLM изучает контекстуализированные встраивания белков, которые фиксируют геномный контекст, а также саму последовательность белка и кодируют биологически значимую и функционально значимую информацию (например, ферментативную функцию, таксономию). Наш анализ моделей внимания показывает, что gLM изучает совместно регулируемые функциональные модули (т.е. опероны). Наши результаты показывают, что неконтролируемое глубокое изучение метагеномного корпуса с помощью gLM является эффективным и многообещающим подходом для кодирования функциональной семантики и регуляторного синтаксиса генов в их геномном контексте и выявления сложных взаимоотношений между генами в геномной области.
Аналогичный контент просматривают другие

Глубокое обучение: новые методы компьютерного моделирования для геномики
Статья 10 апреля 2019 г.
Гёкчен Эраслан, Жига Авсек, … Фабиан Дж. Тайс
Расшифровка функции микробного гена с помощью обработки естественного языка
Статья В открытом доступе 29 сентября 2022 г.
Даниэль Миллер, Ади Стерн и Дэвид Бурштейн

Глубокое обучение предполагает, что экспрессия генов закодирована во всех частях совместно развивающейся взаимодействующей регуляторной структуры генов.
Статья в открытом доступе 01 декабря 2020
Ян Жримец, Кристоф С. Бёрлин, … Алексей Железняк
Введение
Эволюционные процессы приводят к установлению связи между последовательностями, структурой и функцией белка. Полученная в результате парадигма последовательность-структура-функция 1 уже давно обеспечивает основу для интерпретации огромных объемов геномных данных. Недавние достижения в методах прогнозирования структуры белка на основе нейронных сетей (NN) 2 , 3 , а также недавние модели белкового языка (pLM) 4 , 5 , 6 , 7 предполагают, что ориентированные на данные подходы в обучении без учителя могут представлять эти сложные отношения, формируемые эволюция. На сегодняшний день эти модели в основном рассматривают каждый белок как независимую и автономную сущность. Однако белки кодируются в геномах наряду с другими белками, и конкретный геномный контекст, в котором встречается белок, определяется эволюционными процессами, где каждое событие приобретения, потери, дупликации и транспозиции гена подвергается отбору и дрейфу 8 , 9 , 10 . Эти процессы особенно выражены в геномах бактерий и архей, где частые горизонтальные переносы генов (HGT) формируют геномную организацию и разнообразие 11 , 12 . Таким образом, существует присущая эволюционная связь между генами, их геномным контекстом и функцией генов 13 , 14 , 15 , которую можно исследовать путем характеристики закономерностей, возникающих из больших наборов метагеномных данных.
Недавние попытки смоделировать геномную информацию показали прогностическую силу геномного контекста в функции генов 16 и эволюции метаболических признаков 17 в бактериальных и архейных геномах. Однако эти методы представляют гены как категориальные сущности, несмотря на то, что эти гены существуют в непрерывном пространстве, где многомерные свойства, такие как филогения, структура и функции, абстрагированы в их последовательностях. На другом конце спектра представлений были попытки использовать неконтролируемое обучение нуклеотидных последовательностей для прогнозирования уровня экспрессии генов 18 и обнаружения регуляторных мотивов 19 , 20 , 21 . Эти модели в основном обучены и протестированы на геноме человека и ориентированы на прогнозирование регуляции генов, а не их функций. Самые последние попытки использовать разнообразные микробные последовательности для моделирования информации в масштабе генома включают GenSLM 22 , который предварительно обучается на представлениях на уровне кодонов различных последовательностей бактериальных и вирусных генов, а затем точно настраивается на геномах SARS-CoV-2. Чтобы изучить обобщенные межгенные контекстные взаимодействия в биологии, модель должна быть предварительно обучена на 1) разнообразных линиях организмов, 2) богатом и непрерывном представлении генов и 3) более длинных сегментах геномов с множеством генов. Насколько нам известно, не существует метода, который сочетал бы в себе все три аспекта предварительной подготовки для изучения геномной информации в различных биологических линиях (см. краткое изложение предыдущих исследований в дополнительной таблице 1 ).
Чтобы закрыть разрыв между геномным контекстом и последовательностью-структурой-функцией гена, мы разрабатываем модель геномного языка (gLM), которая изучает контекстуальные представления генов. gLM использует встраивания pLM в качестве входных данных, которые кодируют реляционные свойства 4 и структурную информацию 23 генных продуктов. Наша модель основана на архитектуре трансформатора 24 и обучается с использованием миллионов немаркированных метагеномных последовательностей посредством моделирования языка в масках 25 с гипотезой, что ее способность обрабатывать различные части мультигенной последовательности приведет к изучению генов. функциональная семантика и регуляторный синтаксис (например, опероны). Здесь мы сообщаем о доказательствах усвоенных контекстуализированных белковых вложений и моделей внимания, улавливающих биологически значимую информацию. Мы демонстрируем потенциал gLM для прогнозирования функции генов и совместной регуляции, а также предлагаем будущие приложения и направления исследований, включая возможности трансферного обучения gLM.
Полученные результаты
Моделирование геномных последовательностей на языке масок
Языковые модели, такие как представления двунаправленного кодировщика от преобразователей (BERT 25 ), изучают семантику и синтаксис естественных языков, используя неконтролируемое обучение большого корпуса. При моделировании замаскированного языка перед моделью стоит задача восстановить поврежденный входной текст 25 , в котором некоторая часть слов замаскирована. Значительный прогресс в производительности языкового моделирования был достигнут за счет применения архитектуры нейронной сети преобразователя 24 , в которой каждый токен (т. е. слово) может обслуживать другие токены. В этом отличие от сетей с долговременной краткосрочной памятью (LSTM) 26 , которые последовательно обрабатывают токены. Для моделирования геномных последовательностей мы обучили 19-слойную модель трансформера (рис. 1А ; детальный рисунок см. на дополнительном рис. 1 ) на семи миллионах фрагментов метагеномных контигов, состоящих из 15–30 генов из базы данных MGnify 27 . Каждый ген в геномной последовательности представлен вектором из 1280 признаков (встраивание бесконтекстных белков), созданным с использованием ESM2 pLM 23 , объединенным с признаком ориентации (прямой или обратной). Для каждой последовательности 15% генов замаскированы случайным образом, и модель учится предсказывать замаскированную метку, используя геномный контекст. Основываясь на понимании того, что в определенном геномном контексте можно законно обнаружить более одного гена, мы позволяем модели делать четыре различных прогноза, а также предсказывать связанные с ними вероятности (дополнительный рисунок 1 ). Таким образом, вместо того, чтобы предсказывать их среднее значение, модель может аппроксимировать основное распределение множества генов, которые могут занимать геномную нишу. Мы оцениваем производительность модели с помощью показателя псевдоточности, где прогноз считается правильным, если он находится ближе всего к замаскированному белку на евклидовом расстоянии по сравнению с другими белками, закодированными в последовательности (см. «Методы»). Мы проверяем эффективность нашей модели на геноме 28 Escherichia coli K-12, исключив из обучения 5,1% субконтигов MGnify, в которых более половины белков сходны (>70% идентичности последовательностей) с белками E. coli K-12. Важно отметить, что нашей целью не было удаление всех гомологов E. coli K-12 из обучения, что привело бы к удалению подавляющего большинства данных обучения, поскольку многие важные гены являются общими для всех организмов. Вместо этого нашей целью было удалить из обучения как можно больше геномных контекстов (субконтигов), подобных E.coli K-12, что более соответствует цели обучения. gLM достигает 71,9% псевдоточности проверки и 59,2% абсолютной точности проверки (дополнительный рисунок 2 ). Примечательно, что 53,0% прогнозов, сделанных во время проверки, имеют высокую достоверность (с вероятностью прогнозирования > 0,75), а 75,8% прогнозов с высокой достоверностью верны, что указывает на способность gLM изучать показатель достоверности, соответствующий повышенной точности. Мы основываем нашу производительность на двунаправленной модели LSTM, обученной с использованием одной и той же задачи языкового моделирования в одном и том же наборе обучающих данных, где производительность проверки стабилизируется на уровне псевдоточности 28% и абсолютной точности 15% (дополнительный рисунок 2 и дополнительная таблица 2 , обратите внимание, что biLSTM меньше, поскольку не удалось сойтись при масштабировании количества слоев). Мы отказываемся от использования представлений pLM в качестве входных данных для gLM, заменяя их представлениями горячих аминокислот (дополнительная таблица 3 ) и сообщаем о производительности, эквивалентной случайным предсказаниям (псевдоточность 3% и абсолютная точность 0,02%).
Рис. 1: схемы обучения и вывода gLM.
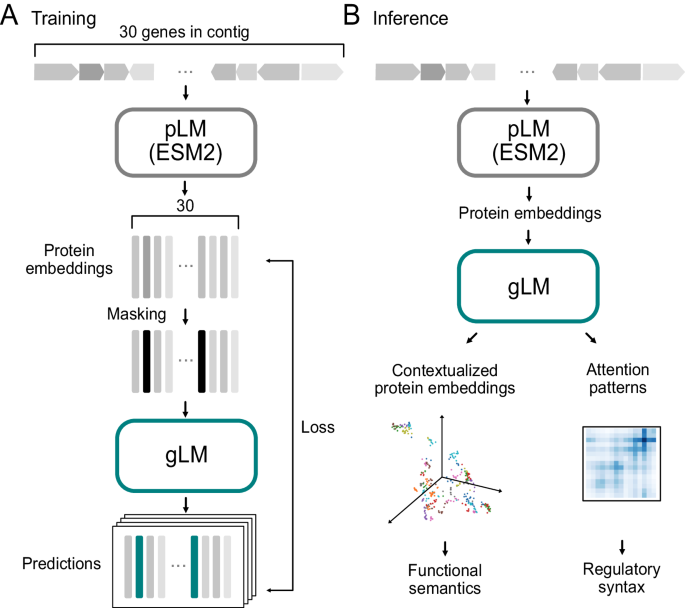
A Для обучения контиги (непрерывные геномные последовательности), содержащие до 30 генов, сначала транслируются в белки, которые впоследствии встраиваются с помощью кодера модели белкового языка (pLM) (ESM2). Маскированные входные данные генерируются путем случайного маскирования с вероятностью 15%, а модель геномного языка (gLM; кодировщик-трансформер) обучается делать четыре прогноза для каждого замаскированного белка с соответствующими вероятностями. Потери при обучении рассчитываются как на основе прогноза, так и на основе вероятностей. B Во время вывода входные данные генерируются из контига с использованием выходных данных ESM2. Контекстуализированные встраивания белков (скрытые слои gLM) и паттерны внимания используются для различных последующих задач. Подробные схемы см. на дополнительном рис. 1 . Исходные данные предоставляются в виде файла исходных данных.
Контекстуализированные встраивания генов отражают семантику генов.
Сопоставление генов с функциями генов в организмах не является однозначно однозначным. Подобно словам в естественном языке, ген может наделять различными функциями 29 в зависимости от его контекста 30 , и многие гены наделяют схожими функциями (т.е. конвергентная эволюция 31 , отдаленная гомология 32 ). Мы использовали gLM для создания контекстуализированных вложений белка с 1280 признаками во время вывода (рис. 1B ) и исследовали «семантическую» информацию, захваченную в этих встраиваниях. Аналогично тому, как слова могут иметь разные значения в зависимости от типа текста, в котором они встречаются (рис. 2А ), мы обнаруживаем, что контекстуализированные белковые внедрения генов, которые появляются в различных средах (биомах), имеют тенденцию группироваться в зависимости от типов биомов. . В нашей обучающей базе данных (MGYP) мы идентифицировали 31 белок, которые встречались более 100 раз и распределялись как минимум по 20 раз в каждом «связанном с хозяином», «экологическом» и «инженерном» биомах в соответствии с обозначением MGnify. Мы обнаружили, что контекстуализированные встраивания белков gLM собирают информацию о биомах для большинства ( n = 21) этих мультибиомных MGYP. Например, ген, кодирующий белок с аннотацией «фактор инициации трансляции IF-1», встречается несколько раз в биомах. В то время как входные данные для gLM (внеконтекстное встраивание белка; представление ESM2) идентичны во всех случаях, выходные данные gLM (встраивание контекстных белков) группируются с типами биомов (рис. 2B ; оценка силуэта = 0,17, см. Остальные 30 мультибиомных MGYP). визуализации на дополнительном рисунке 3 ). Это говорит о том, что различные геномные контексты, которые занимает ген, специфичны для разных биомов, что подразумевает специфичную для биома семантику генов.
Рис. 2: Контекстуальный анализ встраивания белков и сравнение с концепциями моделирования естественного языка.
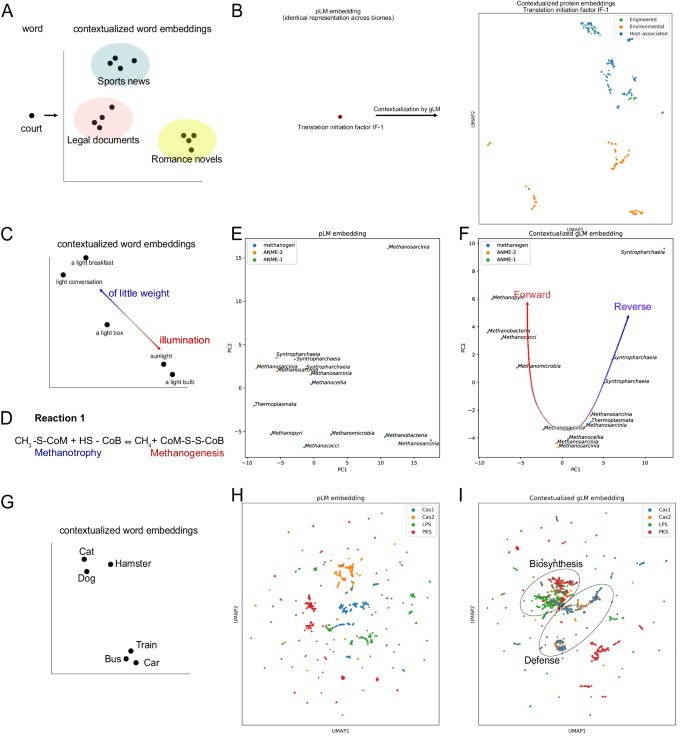
Слово при контекстуализации может быть отображено во вложенное пространство. Для многих слов семантическое значение варьируется в разных типах литературы, и поэтому их контекстуальные вложения группируются в зависимости от типа исходного текста. Рисунок создан для качественной визуализации. B Встраивание входного белка (выходные данные ESM2 и встраивание контекстно-свободного белка) одинаково для всех вхождений белка в базу данных. При контекстуализации с помощью gLM контекстуализированные белковые внедрения одного и того же белка (последний скрытый слой gLM во время вывода) группируются с типом биома, аналогичным типу исходного текста на естественном языке ( A ). Контекстуализацию 30 других мультибиомных MGYP можно найти на дополнительном рисунке 3 . C Значение слова при контекстуализации варьируется в широком диапазоне и может быть неоднозначным даже при контекстуализации (например, двусмысленность). D Реакция 1, осуществляемая комплексом MCR, либо обратная (метанотрофия), либо прямая (метаногенез). E Анализ главных компонентов (PCA) внеконтекстных белковых вложений последовательностей McrA в геномы (общее объясненное отклонение = 0,56), окрашенный в соответствии с метаболической классификацией организма (ANME, метаноген), основанный на предыдущих исследованиях и помеченный таксономией на уровне класса. F PCA контекстуализированных встраиваний McrA (общая объясненная дисперсия = 0,68), где встраивания gLM группируются с направлением реакции 1, которую, скорее всего, осуществит комплекс MCR. G Геометрические отношения между контекстуализированными встраиваниями белков, основанные на семантической близости слов. H Входные (бесконтекстные) встраивания белков Cas1, Cas2, липополисахаридсинтаз (LPS) и поликетидсинтаз (PKS), демонстрирующие кластеризацию на основе структурного сходства и сходства последовательностей. I Кластеризация контекстуальных встраиваний белков, в которых кластеризуются белки фаговой защиты (Cas1 и Cas2) и продукты биосинтеза генов (липополисахаридсинтазы [LPS] и поликетидсинтазы [PKS]). Исходные данные предоставляются в виде файла исходных данных.
Мы исследовали экологически важный пример геномной «полисемии» (множественных значений, придаваемых одному и тому же слову; рис. 2C ) комплекса метил-коэнзим М-редуктазы (MCR). Комплекс MCR способен осуществлять обратимую реакцию (реакция 1 на рис. 2D ), при этом прямая реакция приводит к образованию метана (метаногенез), а обратная — к окислению метана (метанотрофия). Сначала мы исследуем белок McrA (субъединица альфа метил-коэнзима М-редуктазы) в различных линиях ANME (ANaerobic MEthane, окисляющих) и метаногенных геномах архей. Эти археи полифилетичны и занимают определенные экологические ниши. Примечательно, что подобно тому, как семантическое значение слова существует в спектре и слово может иметь несколько семантически соответствующих значений в контексте (рис. 2C ) , комплекс MCR может выполнять различные функции в зависимости от контекста. Предыдущие отчеты демонстрируют способность ANME (в частности, ANME-2) осуществлять метаногенез 33 и метаногенов, осуществляющих окисление метана в специфических условиях роста 34 . Встраивание этих белков в ESM2 без контекста (рис. 2E ) демонстрирует слабую организацию с небольшим разделением между белками McrA ANME-1 и ANME-2. Однако контекстуализированные встраивания белков McrA в gLM (рис. 2F ) демонстрируют четкую организацию: белки McrA ANME-1 образуют плотный кластер, тогда как белки McrA ANME-2 образуют кластер с метаногенами (оценка силуэта после контекстуализации: 0,24; до контекстуализации: 0,027). Эта организация отражает филогенетические взаимоотношения между организмами, в которых обнаружены McrA, а также отчетливые оперонические и структурные различия комплексов MCR в ANME-1 по сравнению с теми, которые обнаружены в ANME-2 и метаногенах 35 . По предложению Шао и др. 35. предпочтительная направленность реакции 1 (рис. 2D ) в ANME-2 и некоторых метаногенах может в большей степени зависеть от термодинамики.
Мы также демонстрируем, что контекстуализированные встраивания gLM более подходят для определения функциональных отношений между классами генов. Аналогично тому, как слова «собака» и «кошка» ближе по значению относительно слов «собака» и «поезд» (рис. 2G ), мы видим закономерность, при которой гены, кодирующие Cas1 и Cas2, кажутся диффузными в нескольких подкластерах в контексте. -свободное пространство для встраивания белка (рис. 2H ) кластер в контекстуализированном пространстве для встраивания (рис. 2I ). Это отражает их сходство функций (например, защита от фагов). Это также продемонстрировано в биосинтетических генах, где гены, кодирующие липополисахаридсинтазу (LPS) и поликетидсинтазу (PKS), группируются ближе друг к другу в контекстуализированном пространстве встраивания, отличном от белков Cas (рис. 2I ). Мы количественно оцениваем эту закономерность с помощью более высокого силуэтного показателя, измеряющего фаговую защиту и биосинтетическое разделение генов (представление gLM: 0,123 ± 0,021, представительство pLM: 0,085 ± 0,007; парный t-критерий, t-статистика: 5,30, двусторонний, значение p = 0,0005). , n = 10). Таким образом, контекстуализированные встраивания белков способны фиксировать реляционные свойства, подобные семантической информации 36 , где гены, кодирующие белки, которые более схожи по своим функциям, обнаруживаются в схожих геномных контекстах.
Чтобы количественно оценить прирост информации в результате обучения преобразователя геномным контекстам, мы сравниваем результаты кластеризации в 2B, F и I с кластеризацией, проводимой на (суб)усредненных встраиваниях pLM (дополнительный рисунок 4 ). Объединив вложения pLM по среднему значению для данного подконтига, мы можем обобщить контекстную информацию в качестве простой базовой линии. Мы сообщаем о наиболее последовательной кластеризации (более высокие оценки силуэта) встраивания gLM по сравнению с pLM, усредненным по контигам, для всех трех анализов (значения см. в подписях к дополнительным рисункам 4 ). Мы демонстрируем, что модель преобразователя gLM изучает представления, которые коррелируют с биологической функцией, которые не фиксируются наивной базовой линией.
Характеризуя неизвестное
Метагеномные последовательности содержат множество генов с неизвестными или родовыми функциями, а некоторые настолько расходятся, что не имеют достаточного сходства последовательностей с аннотированной частью базы данных 37 . В нашем наборе данных из 30,8 M белковых последовательностей 19,8% не удалось связать ни с одной известной аннотацией (см. «Методы»), а 27,5% не удалось связать с какими-либо известными доменами Pfam с использованием недавнего подхода глубокого обучения (ProtENN 38 ). Понимание функциональной роли этих белков в их организменном и экологическом контексте остается серьезной проблемой, поскольку большинство организмов, содержащих такие белки, трудно культивировать, а лабораторная проверка часто имеет низкую производительность. В микробных геномах белки, выполняющие сходные функции, обнаруживаются в сходных геномных контекстах из-за селективного давления, создаваемого функциональными взаимоотношениями (например, белок-белковыми взаимодействиями, совместной регуляцией) между генами. Основываясь на этом наблюдении, мы предположили, что контекстуализация предоставит более богатую информацию, которая приблизит распределение неаннотированных генов к распределению аннотированных генов. Мы сравнили распределения неаннотированных и аннотированных фракций белков в нашем наборе данных, используя контекстно-свободные встраивания pLM и контекстуализированные встраивания gLM. Мы обнаружили статистически значимое меньшее расхождение между распределениями неаннотированных и аннотированных генов во встраиваниях gLM по сравнению с встраиваниями pLM (парный t-критерий расхождений Кульбака-Лейблера, статистика t-критерия = 7,61, двусторонний, значение p < 1e-4 , n = 10 (см. «Методы выборки и расчета метрик»). Это предполагает больший потенциал использования встраивания gLM для переноса проверенных знаний о культивируемых и хорошо изученных штаммах (например, E. coli K-12) в практически некультивируемое пространство метагеномных последовательностей. Геномный контекст, наряду с молекулярной структурой и филогенией, по-видимому, является важной информацией, которую необходимо абстрагировать, чтобы эффективно представлять последовательности и обнаруживать скрытые связи между известными и неизвестными частями биологии.
Контекстуализация улучшает прогнозирование функций ферментов
Чтобы проверить гипотезу о том, что геномный контекст белков можно использовать для прогнозирования функций ферментов, мы оценили, как контекстуализация может улучшить выразительность представлений белков для прогнозирования функций ферментов. Сначала мы создали специальный набор данных MGYP-EC, в котором данные обучения и испытаний были разделены с 30% идентичностью последовательностей для каждого класса EC (см. «Методы»). Во-вторых, мы применяем линейный зонд (LP) для сравнения выраженности представлений на каждом слое gLM с маскированием запрашиваемого белка и без него (дополнительный рисунок 5 ). Маскируя запрашиваемый белок, мы можем оценить способность gLM изучать функциональную информацию о данном белке только из его геномного контекста, без распространения информации из встраивания pLM белка. Мы заметили, что большая часть контекстной информации, относящейся к ферментативным функциям, усваивается в первых шести слоях gLM. Мы также демонстрируем, что сама по себе контекстная информация может прогнозировать функцию белка с точностью до 24,4 ± 0,8%. Напротив, без маскировки gLM может включать информацию, присутствующую в контексте исходной информации pLM для каждого запрашиваемого белка. Мы наблюдали увеличение выразительности встраивания gLM и в более мелких слоях, с точностью до 51,6 ± 0,5% в первом скрытом слое. Это означает увеличение на 4,6 ± 0,5% по сравнению с точностью контекстно-свободного прогнозирования pLM (рис. 3A ) и увеличение средней точности на 5,5 ± 1,0% (рис. 3C ). Таким образом, мы демонстрируем, что информация, которую gLM узнает из контекста, ортогональна информация, собранная при внедрении PLM. Мы также наблюдали снижение экспрессивности информации о функциях ферментов в более глубоких слоях gLM; это согласуется с предыдущими исследованиями LLM, где более глубокие уровни специализируются на задаче предварительного обучения (предсказание маскированных токенов), и согласуется с предыдущими исследованиями LLM, где наиболее эффективный уровень зависит от конкретных последующих задач 39 . Наконец, чтобы дополнительно изучить выраженность этих представлений, мы сравнили прирост баллов F1 для каждого класса (рис. 3B ). Мы наблюдаем статистически значимые различия в показателях F1 (t-критерий, двусторонний, скорректированное по Бенджамини/Хохбергу значение p <0,05, n = 5) между двумя моделями в 36 из 73 классов ЕС с более чем десятью образцами в тестовом наборе. . Большинство (27 из 36) статистических различий привели к улучшению показателя F1 у LP, обученного на представлениях gLM.
Рис. 3: Контекстуализация функции гена.
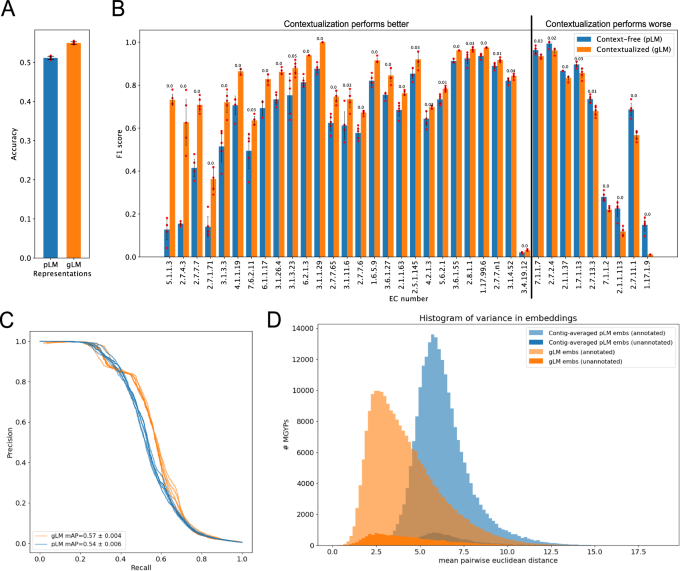
Точность классификации чисел Комиссии по ферментам линейных зондов (EC) для представлений pLM (ESM2) и представлений gLM (1-й скрытый слой). Данные представлены как средние значения +/- стандартное отклонение по пяти техническим повторам. B Сравнение показателей F1 статистически значимых (t-критерий, двусторонний, скорректированное значение p Бенджамини/Хохберга <0,05, технические повторы = 5) различий в эффективности линейных датчиков числа ЕС на основе pLM и gLM. Классы EC упорядочены от наибольшего выигрыша при контекстуализации слева до наибольшего убытка при контекстуализации справа. Данные представлены как средние значения +/- стандартное отклонение. Скорректированное значение p (с двумя значащими цифрами) для каждого класса указано над столбцами. C Кривые Precision-Recall для линейных датчиков числа EC на основе pLM и gLM. D Гистограмма дисперсии (количество ячеек = 100), рассчитанная с использованием контекстуализированных вложений (gLM; оранжевый) и усредненных по контигам встраиваниях pLM (синий) MGYP, которые встречаются в базе данных не менее 100 раз. Гистограммы для неаннотированной и аннотированной части MGYP строятся отдельно, а столбцы не складываются друг на друга. Аннотированные примеры в длинном правом хвосте включают фаговые белки и транспозазы, что отражает их способность к самомобилизации (см. аннотации десятков наиболее вариантов генов в дополнительной таблице 4 ). Исходные данные предоставляются в виде файла исходных данных.
Частота горизонтального переноса соответствует дисперсии встраивания геномного контекста.
Ключевым процессом, который формирует организацию и эволюцию микробного генома, является горизонтальный перенос генов (HGT). Таксономический диапазон, в котором гены распределяются по древу жизни, зависит от их функции и селективного преимущества, которое они получают в различных средах. Относительно мало известно о специфичности геномной области, в которую ген переносится на филогенетические расстояния. Мы исследовали дисперсию встраивания gLM для белков, которые встречаются в базе данных не менее ста раз. Вариация геномных контекстов, изученных с помощью gLM, рассчитывается путем взятия случайной выборки из 100 вхождений и последующего расчета средних попарных расстояний между сотнями встраивания gLM. Мы проводим такую независимую случайную выборку и расчет расстояния десять раз для каждого гена, а затем вычисляем среднее значение. В качестве базовой линии мы рассчитываем дисперсию вложений pLM, усредненных по субконтигам, используя тот же метод выборки, чтобы сравнить информацию, полученную в результате обучения gLM. Наши результаты показывают, что дисперсии геномного контекста, полученные с помощью gLM, имеют более длинный правый хвост (эксцесс = 1,02, асимметрия = 1,08) по сравнению с усредненной по контигам базовой линией pLM, которая имеет более пик (эксцесс = 2,2, асимметрия = 1,05) (рис. 3D ). Примечательно, что большинство контекстно-вариантных генов в правом хвосте распределения контекстных отклонений, изученных с помощью gLM (оранжевый), включали фаговые гены и транспозазы, что отражает их способность к самомобилизации. Интересно, что мы не обнаружили никаких фаговых генов в крайнем правом хвосте усредненного по контигам распределения дисперсии встраивания pLM (синий), хотя мы обнаружили гены, участвующие в транспозиции (дополнительная таблица 4 ). Отклонения геномного контекста, полученные с помощью gLM, можно использовать в качестве показателя частоты горизонтального переноса и для сравнения влияния приспособленности геномного контекста на эволюционную траекторию (например, поток генов) генов, а также для выявления недостаточно охарактеризованных и функциональных мобильных транспонируемых генов. элементы.
Внимание трансформера захватывает опероны
Механизм 24 внимания преобразователя моделирует парное взаимодействие между различными токенами во входной последовательности. Предыдущие исследования моделей внимания моделей преобразователей в обработке естественного языка (НЛП) 39 показали, что разные головы, по-видимому, специализируются на синтаксических функциях. Впоследствии было показано , что различные головы внимания в pLM 40 коррелируют со специфическими структурными элементами и функциональными сайтами в белке. Для нашего gLM мы предположили, что особое внимание сосредоточено на обучающих оперонах, «синтаксической» особенности, выраженной в микробных геномах, где множественные гены со связанными функциями экспрессируются в виде одиночных полицистронных транскриптов. Опероны широко распространены в геномах бактерий, архей и их вирусов и редко встречаются в геномах эукариот. Для проверки мы использовали базу данных 41 оперонов E.coli K-12, состоящую из 817 оперонов. gLM содержит 190 головок внимания на 19 слоях. Мы обнаружили, что головы в более мелких слоях больше коррелировали с оперонами (рис. 4A , дополнительный рисунок 6) , при этом исходные показатели внимания в 7-й голове 2-го слоя [L2-H7] линейно коррелировали с оперонами с коэффициентом корреляции 0,44 (ро Пирсона, Бонферрони скорректировал значение p <1E-5) (рис. 4B ). Далее мы обучили классификатор логистической регрессии (предиктор оперонов), используя все модели внимания во всех головах. Наш классификатор предсказывает наличие оперонической связи между парой соседних белков в последовательность с высокой точностью (средняя средняя точность = 0,775 ± 0,028, пятикратная перекрестная проверка) (рис. 4C ). Мы определяем эту производительность, обучая предиктор оперона на абляции gLM на основе представления одной горячей аминокислоты (среднее значение средняя точность = 0,426 ± 0,015, пятикратная перекрестная проверка; дополнительная таблица 3 ), которая учится на основе информации об ориентации и совместном возникновении, но не может в полной мере использовать богатое представление генов.
Рис. 4: Анализ внимания.
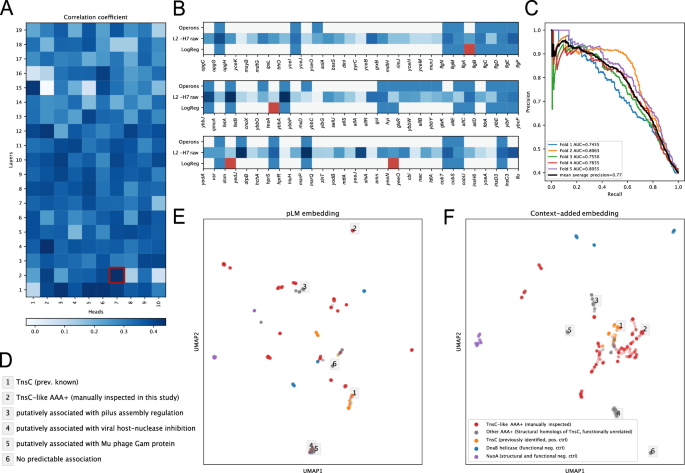
Коэффициенты корреляции (ро Пирсона) между головами внимания по слоям и оперонам. Более темный цвет соответствует более сильной корреляции с ранее идентифицированными оперонами. Паттерны внимания второго слоя-седьмой головы [L2-H7] наиболее сильно коррелируют с оперонами. B Три случайных примера контигов и предсказанные оперонные отношения между соседними белками. Белки перечислены в том порядке, в котором они закодированы в контиге. Основная истина Опероны E.coli K-12 (верхний ряд), необработанные показатели внимания в головке внимания [L2-H7] наиболее коррелируют с оперонами (средний ряд) и прогноз логистической регрессии с использованием всех головок внимания (последний ряд), где ложноположительные прогнозы (или, возможно, неверно аннотированные основные истины в случае жгутиковых белков в первом примере) отмечены красным. C Кривые точности и отзыва с пятикратной перекрестной проверкой логистической регрессии, обученные с использованием всех оперонов и головок внимания. D Ассоциации регуляторов AAA+ , охарактеризованные с помощью предсказания оперонов на основе внимания ( расширенная фигура 11A ), соответствующих примерам, отмеченным на панелях E и F. E ESM2 генерирует встраивания входных белков белков-регуляторов AAA+, которые являются структурными гомологами TnsC (серый и красный; с использованием Foldseek 60 ). Структурные гомологи TnsC с подтвержденным участием в Tn7-подобных транспозонах при ручной проверке были обозначены как «TnsC-подобные AAA+ (проверка вручную)» и окрашены в красный цвет. Другие белки MGYP, отмеченные как «TnsC» в базе данных UniRef90 (оранжевый), были добавлены в качестве положительного контроля функции TnsC. NuoA (субъединица А НАДН-хинон-оксидоредуктазы ; фиолетовый) добавляли в качестве структурного и функционального отрицательного контроля. Геликазы DnaB (синие) были добавлены в качестве функционального отрицательного контроля, поскольку эти белки имеют складки, аналогичные TnsC, но не связаны с транспозицией. F Комбинированный входной белок и контекстные встраивания генов на панели E. Эти внедрения создаются посредством объединения внедрений pLM (ESM2) и внедрений контекста (последний уровень gLM). Отрицательные контроли (геликазы NuoA и DnaB) образуют отдельные кластеры как в E , так и в F. Пронумерованные метки в серых прямоугольниках обозначают белки AAA+ с предсказаниями различных функциональных ассоциаций, перечисленных на панели D и на дополнительном рисунке 7 . Кластеризация вложений на основе необработанного расстояния показана на дополнительном рисунке 8 . Исходные данные предоставляются в виде файла исходных данных.
Контекстная зависимость функций регулятора ААА+ в сложных генетических системах
Понимание функциональной роли регуляторного белка в организме остается сложной задачей, поскольку одна и та же белковая складка может выполнять разные функции в зависимости от контекста. Например, белки AAA+ (АТФазы, связанные с разнообразной клеточной активностью) используют химическую энергию гидролиза АТФ для обеспечения разнообразных механических клеточных функций 42 . Однако регуляторы AAA+ могут также играть совершенно разные, широкие функциональные роли в зависимости от своих партнеров по клеточному взаимодействию: от деградации белка и репликации ДНК до транспозиции ДНК. Одним особенно интересным примером является белок TnsC, который регулирует активность вставки ДНК 43 в Tn7-подобные транспозонные системы. Многочисленные биоинформационные усилия были сосредоточены на открытии ранее неохарактеризованных транспозонов посредством метагеномного поиска 44 и поиска последовательностей собранных геномов 45 , направленных на идентификацию подходящих гомологов для приложений редактирования генома 46 . Чтобы проверить, могут ли разработанные здесь методы идентифицировать Tn7-подобные системы транспозиции, а также отличать их от других функциональных контекстов, мы исследовали контекстуализированную семантику структурных гомологов TnsC в базе данных MGnify. Без контекстуализации не возникает кластеризации со связанной с ней транспозазной активностью (коэффициент расхождения KL = 1,03; см. Методы расчета этого показателя, рис. 4E ). Однако с добавленной контекстуализацией ранее идентифицированный TnsC (оранжевый) и проверенный вручную структурный гомолог TnsC (красный, помечен как «TnsC-подобный») кластеризуется вместе (коэффициент расхождения KL = 0,38; рис. 4F ; см. дополнительные рис. 7B, C). для сравнения с базовыми показателями pLM только для gLM и усредненными по контигам). Мы дополнительно проверяем эту визуализацию, используя встраивание кластеризации на основе расстояний (дополнительный рисунок 8 ). Многие структурные гомологи TnsC не участвовали в транспозиции, и это отражено в отдельных кластерах серых точек данных вдали от известных TnsC (оранжевые) и TnsC-подобных структурных гомологов (красные) на фиг. 4F . Эти кластеры представляют собой разнообразную и контекстно-зависимую регуляционную активность AAA+, которую невозможно предсказать ни по структуре, ни по исходной последовательности. Мы предсказали опероническую связь между этими регуляторами AAA+ и соседними с ними генами и обнаружили, что многие из них находятся в оперонических отношениях с генными модулями разнообразной функции, включая сборку пилуса и ингибирование нуклеазы вирусного хозяина (рис. 4D , дополнительная фиг. 7A ). В некоторых случаях запрашиваемые белки AAA+, по-видимому, не находились в оперонной ассоциации с соседними белками, что позволяет предположить, что некоторые белки AAA+ с меньшей вероятностью будут функционально связаны со своими соседями, чем другие (дополнительная фигура 7A , пример 6). Используя этот пример регуляторов AAA+, мы показываем, что объединение контекстуализированных встраиваний белков и взаимодействия оперонов, основанного на внимании, может обеспечить важный путь для изучения и характеристики функционального разнообразия регуляторных белков.
gLM предсказывает паралогию межбелковых взаимодействий
Белки в организме находятся в комплексах и физически взаимодействуют друг с другом. Недавние достижения в предсказании белок-белковых взаимодействий (PPI) и исследованиях структурных комплексов в значительной степени были обусловлены идентификацией интерологов (консервативных PPI в организмах) и коэволюционных сигналов между остатками 47 . Однако различение паралогов от ортологов (также известное как проблема «сопоставления паралогов») в расширяющемся наборе данных последовательностей остается вычислительной задачей, требующей запросов по всей базе данных и/или филогенетического профилирования. В случаях, когда в организме обнаруживаются множественные взаимодействующие пары (например, гистидинкиназы (HK) и регуляторы ответа (RR)), предсказание взаимодействующих пар особенно сложно 48 . Мы рассудили, что gLM, хотя и не обучен непосредственно этой задаче, возможно, изучил отношения между паралогами и ортологами. Чтобы проверить эту возможность, мы использовали хорошо изученный пример взаимодействующих паралогов (ModC и ModA, рис. 5А ), которые образуют транспортный комплекс ABC. Мы запросили gLM, чтобы предсказать встраивание взаимодействующей пары, не учитывая никакого контекста, кроме белковой последовательности ModA или ModC. Мы обнаружили, что без какой-либо тонкой настройки gLM работает как минимум на порядок лучше, чем можно было ожидать по случайности (см. «Методы»). В частности, для 398 из 2700 взаимодействующих пар gLM делает прогнозы, которые принадлежат тому же кластеру (50% идентичности последовательности, n = 2100 кластеров), что и истинная метка, а в 73 парах gLM прогнозирует метку, которая наиболее близка к точная взаимодействующая пара (смоделированное случайное совпадение = 1,6 ± 1,01, n = 10) (рис. 5B ). Важно отметить, что при рассмотрении только прогнозов с очень высокой степенью достоверности (правдоподобие прогноза> 0,9, n = 466) gLM способен сопоставлять паралоги с повышенной точностью на 25,1%. Когда паралоги правильно спарены, gLM более уверен в предсказании (средняя достоверность правильного предсказания = 0,79, средняя достоверность по всем предсказаниям = 0,53), в то время как менее определённые предсказания либо выходят за рамки распределения, либо ближе к среднему значению меток (рис. 5С ) . Мы объясняем часть неточностей в прогнозировании тем фактом, что gLM не был обучен задаче прогнозирования замаскированного гена, учитывая только один ген в качестве геномного контекста, хотя мы ожидаем, что производительность улучшится с расширением диапазона длины обучающей последовательности и более точным -настройка модели специально для задачи «сопоставления паралогов».
Рис. 5: Потенциал для трансферного обучения.
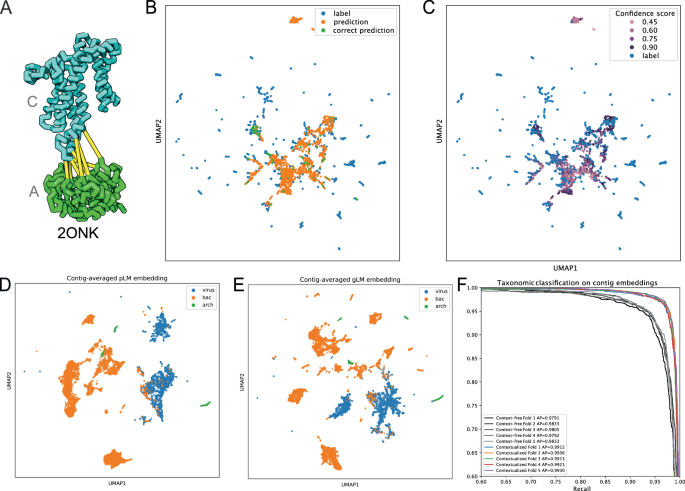
Взаимодействие ModA и ModC (структура банка данных белков 2ONK) 47 B UMAP-проекция предсказаний (оранжевый) и меток (синий) паралогов (ModAC показан на рисунке A), где правильные предсказания окрашены в зеленый цвет. C Прогнозируемые вложения окрашиваются в зависимости от прогнозируемой достоверности. Прогнозы вне распределения и прогнозы, близкие к среднему значению, обычно имеют меньшую достоверность, тогда как правильные прогнозы имеют более высокую достоверность. D, E Случайные 30-генные контиги из репрезентативных бактериальных («bac») и архейных («arch») геномов и эталонных вирусных («vir») геномов были встроены посредством встраивания белков ESM2 со средним пулом (контекстно-свободные встраивания контигов, D , E). ) и путем среднего объединения последнего скрытого слоя gLM (контекстуализированных вложений контигов, E ). F Микроусредненные кривые точности-памяти и средние точности для классификаторов логистической регрессии, обученных с использованием контекстно-свободных вложений контигов (серые линии) и контекстуальных вложений контигов (цветные линии) для задачи классификации таксономии на уровне класса. Каждая линия представляет собой складку в стратифицированной k-кратной перекрестной проверке (k = 5). Таксономия уровня класса для каждого контига показана на дополнительных рисунках 9A, B , а матрицы путаницы для классификаторов логистической регрессии показаны на дополнительных рисунках 9C, D. Исходные данные предоставляются в виде файла исходных данных.
Контекстуализированные встраивания контигов и потенциал для трансферного обучения
Контекстуализированные встраивания белков кодируют взаимосвязь между конкретным белком и его геномным контекстом, сохраняя последовательную информацию внутри контига. Мы предположили, что эта контекстуализация добавляет биологически значимую информацию, которую можно использовать для дальнейшей характеристики мультигенных геномных контигов. Здесь мы определяем контекстуализированное встраивание контига как скрытый слой со средним пулом по всем белкам в субконтиге, а контекстно-свободное встраивание контига как встраивание белка ESM2 со средним пулом по всей последовательности (см. методы). Оба вложения состоят из 1280 функций. Мы проверяем нашу гипотезу, исследуя способность каждого из этих вложений линейно отличать вирусные последовательности от бактериальных и архейных субконтигов. В наборах метагеномных данных таксономическая идентичность собранных последовательностей должна быть выведена post hoc, поэтому идентификация вирусных последовательностей проводится на основе присутствия вирусных генов и вирусных геномных сигнатур 49 . Однако такая задача классификации остается сложной, особенно для более мелких фрагментов контигов и менее охарактеризованных вирусных последовательностей. Здесь мы выбрали случайные 30-белковые субконтиги из репрезентативной базы данных геномов бактерий и архей и эталонных вирусных геномов в NCBI и визуализировали их бесконтекстные встраивания контигов (рис. 5D ) и контекстуализированные встраивания контигов (рис. 5E ). Мы наблюдали большее разделение и таксономические кластеры как на уровне домена, так и на уровне классов (дополнительная фигура 9 ) , что позволяет предположить, что таксономическая подпись усиливается за счет кодирования скрытых отношений между белками. Это дополнительно подтверждается обучением классификатора логистической регрессии на контекстуальных и контекстуализированных вложениях контигов для таксономии уровня класса (дополнительные рисунки 9A, B ), где мы видим статистически значимое улучшение средней точности (рис. 5F , см. дополнительный рисунок). 7C , D для матриц неточностей). Это подчеркивает биологическую важность относительного положения белка в геноме и его связи с геномным контекстом, а также указывает на то, что эта информация может быть эффективно закодирована с использованием gLM. Контекстуализированные встраивания контигов открывают возможности для трансферного обучения, выходящие за рамки предсказания вирусных последовательностей, такие как улучшенное объединение метагеномно собранного генома (MAG) и коррекция сборки.
Обсуждение
Беспрецедентное количество и разнообразие метагеномных данных в сочетании с достижениями в области глубокого обучения открывают захватывающие возможности для создания моделей, способных изучать скрытые закономерности и структуры биологических систем. Такие модели основаны на концептуальных и статистических основах, разработанных биологами-эволюционистами за последнее столетие. Обладая способностью абстрагировать гораздо большие объемы данных, эти модели могут распутать чрезвычайную сложность геномов организмов и их закодированных функций; это ключевой шаг в углублении нашего понимания биологических процессов. Представленная здесь работа демонстрирует и подтверждает концепцию моделирования геномного языка. Наша реализация моделирования замаскированного геномного языка иллюстрирует возможность обучения такой модели и предоставляет доказательства того, что биологически значимая информация фиксируется в изученных контекстуальных встраиваниях и дает значимые интерпретации моделей внимания. Мы показываем, что gLM можно использовать для различных последующих задач, включая прогнозирование функций ферментов, прогнозирование оперонов, сопоставление паралогов и прогнозирование таксономии контигов. Кроме того, мы демонстрируем способность gLM освещать контекстную зависимость функций в рамках структурной гомологии и гомологии последовательностей на примере регуляторов AAA +. В совокупности gLM представляет собой весьма многообещающее направление для интерпретации биологии, и мы предлагаем ключевые области для дальнейшего развития: во-первых, архитектура преобразователя доказала свою эффективность в эффективном масштабировании; Как при обработке естественного языка 50, так и при обработке белкового языка 23 было показано, что увеличение количества параметров в модели вместе с размером набора обучающих данных приводит к значительному улучшению производительности и возможности обобщения. Наша модель состоит из ~1B параметров, что как минимум на величину меньше по сравнению с современными pLM. При дальнейшей настройке и масштабировании гиперпараметров мы ожидаем повышения производительности модели. Во-вторых, наша модель в настоящее время использует встраивания pLM для представления белков на входе. Эти встраивания генерируются путем объединения скрытых состояний на уровне аминокислотных остатков по всей белковой последовательности, и поэтому информация, специфичная для остатков, и эффекты синонимических мутаций, вероятно, скрыты. Будущие итерации модели могут использовать необработанные встраивания на уровне остатков или кодонов в качестве входных данных, чтобы позволить моделировать коэволюционные взаимодействия остатков к остаткам между белками и влияние синонимических мутаций на функцию гена. В-третьих, задача реконструкции встраивания замаскированных белков требует моделирования распределения по возможным встраиваниям; наш метод аппроксимирует это распределение, используя фиксированное количество прогнозов. Будущая работа может улучшить эту ситуацию, используя генеративный подход, такой как модель диффузии или GAN. Это может обеспечить лучшую точность прогнозирования и большую возможность обобщения невидимых наборов данных. В-четвертых, добавление небелковых модальностей (например, некодирующих регуляторных элементов) в качестве входных данных в gLM может также значительно улучшить представление gLM данных о биологических последовательностях и может изучить функцию и регуляцию белка, обусловленную другими модальностями 51 . Наконец, наша модель была обучена в основном на геномах бактерий, архей и вирусов, поэтому то, как этот метод можно адаптировать для геномов эукариот, особенно тех, которые имеют обширные межгенные области, еще предстоит изучить.
Одним из наиболее мощных аспектов языковых моделей на основе преобразователей является их потенциал для переноса обучения и тонкой настройки. Мы протестировали некоторые возможности gLM и успешно показали, что биологическую информацию более высокого порядка, включая функцию и регуляцию генов, можно изучить с помощью геномных последовательностей. Наши результаты подчеркивают важность контекстуализации биологических данных, особенно когда мы масштабируем наши усилия по моделированию от биомолекул до целых организмов. Мы предлагаем следующие многообещающие будущие направления применения gLM для продвижения биологических исследований. 1) Перенос обучения на основе признаков для прогнозирования функций белков (например, термин Gene Ontology [GO]), особенно с ограниченной последовательностью и структурной гомологией. 2) Точная настройка gLM для задачи прогнозирования межбелкового взаимодействия. 3) Использование функций gLM для кодирования геномного контекста в качестве дополнительных входных данных для улучшенных и контекстуализированных прогнозов структуры белка. В заключение, моделирование геномного языка является мощным инструментом для беспристрастного сжатия важной биологической информации из полных метагеномных последовательностей. В сочетании с достижениями в области секвенирования длительного чтения мы ожидаем резкого повышения качества, количества и разнообразия входных данных. Моделирование геномного языка открывает возможность преодолеть разрыв между атомной структурой и функцией организма и тем самым приближает нас к моделированию биологических систем и, в конечном итоге, к точному манипулированию биологией (например, редактированию генома, синтетической биологии).
Методы
База данных последовательностей
Геномный корпус был создан с использованием набора данных MGnify 27 (выпущен 6 мая 2022 г. и загружен 7 июня 2022 г.). Сначала геномные контиги с более чем 30 генами были разделены на 30 непересекающихся генных субконтигов, в результате чего в общей сложности получилось 7 324 684 субконтига с длиной от 15 до 30 генов (субконтиги длиной менее 15 генов были удалены из набора данных). Мы выбрали 30 в качестве максимальной длины контекста, потому что, хотя более длинный контекст приводит к более высокой производительности моделирования (дополнительный рисунок 10A ), 67% необработанных контигов MGnify с > 15 генами имели длину =<30 генов (дополнительный рисунок 10B ), и поэтому увеличение длины контекста сверх 30 привело бы ко многим примерам с заполнением (снижение эффективности вычислений). Каждый ген в подконтиге был картирован на репрезентативную белковую последовательность (репрезентативный MGYP) с использованием mmseqs/linclust 52 , с пороговыми значениями охвата и идентичности последовательностей, установленными на уровне 90% (предварительно рассчитанными в базе данных MGnify), в результате чего в общей сложности было получено 30 800 563 репрезентативных MGYP. . Каждый репрезентативный MGYP был представлен встраиванием белка с 1280 признаками, созданным путем объединения среднего значения последнего скрытого слоя модели ESM2 23 «esm2_t33_650M_UR50D». Из-за ограничения памяти при вычислении вложений для очень длинных последовательностей 116 последовательностей MGYP длиной более 12290 аминокислот были усечены до 12290 аминокислот. Вложения ESM2 были нормализованы (путем вычитания среднего значения каждой функции и деления на ее стандартное отклонение) и обрезаны таким образом, чтобы все функции находились в диапазоне от -10 до 10, чтобы улучшить стабильность обучения. Небольшую часть (0,4%) генов не удалось сопоставить с репрезентативным MGYP, и поэтому информацию о соответствующей последовательности нельзя было получить с сервера MGnify; этим последовательностям был присвоен вектор признаков из 1280 единиц. Для каждого гена в подпоследовательности мы добавили признак ориентации гена к стандартизированному встраиванию белка MGYP, где 0,5 обозначает «прямую» ориентацию относительно направления секвенирования, а -0,5 обозначает «обратную» ориентацию. Таким образом, каждый ген был представлен в нашем корпусе 1281 вектором признаков.
Архитектура и обучение gLM
gLM был построен на основе реализации архитектуры трансформатора RoBERTa 53 . gLM состоял из 19 слоев со скрытым размером 1280 и десятью заголовками внимания на слой с встраиванием относительного положения («relative_key_query») 54 . Для обучения 15% токенов (генов) в последовательности (субконтиге) были случайным образом замаскированы до значения -1. Затем мы поставили перед моделью задачу спрогнозировать метку замаскированного токена, где метка состоит из вектора из 100 признаков, состоящего из 99 основных компонентов, отбеленных PCA (объяснимая дисперсия = 89,7%. Дополнительный рисунок 11 ) соответствующее встраивание белка ESM2, объединенное с его особенностью ориентации. Уменьшение размерности меток с использованием PCA повысило стабильность обучения. В частности, gLM проецирует последнее скрытое состояние модели в четыре вектора по 100 признаков и четыре соответствующих значения правдоподобия, используя линейный слой. Общие потери рассчитываются по следующей формуле. ( 1 ).
$$ {{{{{ m{MSE}}}}}}({{{{{ m{ближайший}}}}}}; {{{{{ m{прогноз}}}}}}} ,{{{{{ m{label}}}}}}) + alpha , * , {{{{{ m{CrossEntropyLoss}}}}}}}left({{{{{ m{правдоподобия}}}}}},{{{{{ m{ближайший}}}}}}; {{{{{ m{прогноз}}}}}}; {{{{{ m{index}}}}}} ight)$$
(1)
Ближайший прогноз определяется как прогноз, ближайший к метке, рассчитанный по расстоянию L2. Положим ? = 1e-4. Обучение gLM проводилось с половинной точностью с размером пакета 3000 с распределенным распараллеливанием данных на четырех графических процессорах NVIDIA A100 за 1 296 960 шагов (560 эпох), включая 5 000 шагов прогрева для достижения скорости обучения 1e-4 с помощью оптимизатора AdamW 55 .
Метрика производительности и проверка
Чтобы оценить качество модели и ее обобщаемость за пределами набора обучающих данных, мы используем метрику псевдоточности, где мы считаем прогноз «правильным», если он ближе всего по евклидову расстоянию к метке замаскированного гена относительно другие гены субконтига. Расчет псевдоточности описан в уравнении. ( 2 ).
$$ {{{{{ m{псевдо}}}}}} ; {{{{{ m{accuracy}}}}}} = frac{{# }{{{{{ m{count}}}}}}({{{{{ m{argmin }}}}}}({{{{{ m{dist}}}}}}({{{{{ m{prediction}}}}}},{{{{{ m{labels}} }}}} ; {{{{{ m{in}}}}}} ; {{{{{ m{subcontig}}}}}}))=={{{{{ m{ index}}}}}}({{{{{ m{masked}}}}}} ; {{{{{ m{gene}}}}}}))} {{ # }{{ {{{ m{маскировано}}}}}} ; {{{{{ m{гены}}}}}}}$$
(2)
Мы решили проверить нашу метрику и последующий анализ на наиболее аннотированном на сегодняшний день геноме: E.coli K-12 56 . Чтобы удалить как можно больше субконтигов, подобных E.coli K-12, из набора обучающих данных, мы удалили 5,2% субконтигов, в которых более половины генов были похожи на > 70% (рассчитано с помощью поиска mmseqs2 52 ) по аминокислотной последовательности. к генам E.coli K-12. Мы проверяем нашу метрику псевдоточности, вычисляя абсолютную точность генома E.coli K-12, для которого каждый ген был последовательно замаскирован (уравнение ( 3 ))
$$ {{{{{ m{абсолютный}}}}}} ; {{{{{ m{accuracy}}}}}} = frac{ { # }{{{{{ m{count}}}}}}({{{{{ m{argmin }}}}}}({{{{{ m{dist}}}}}}({{{{{ m{prediction}}}}}},{{{{{ m{all}} }}}} ; {{{{{ m{гены}}}}}} ; {{{{{ m{in}}}}}} ; {{{{{ m{E} }}}}} . {{{{{ m{coli}}}}}} ; {{{{{ m{K}}}}}} - 12))=={{{{{ rm{index}}}}}}({{{{{ m{masked}}}}}} ; {{{{{ m{gene}}}}}}}))}{ { # } {{{{{ m{гены}}}}}} ; {{{{{ m{in}}}}}} ; {{{{{ m{E}}}}}} . {{{{{ m{coli}}}}}} ; {{{{{ m{K}}}}}} - 12}$$
(3)
Контекстуализированный расчет и визуализация внедрения
Контекстуализированное встраивание гена в белок рассчитывается путем сначала ввода субконтига из 15-30 генов, содержащего интересующий ген, а затем выполнения вывода по субконтигу с использованием обученного gLM без маскировки. Затем мы используем последний скрытый слой модели, соответствующий гену, в качестве встраивания, состоящего из 1280 признаков.
Генная аннотация
Гены были аннотированы с использованием Diamond v2.0.7.145 57 по базе данных UniRef90 58 с пороговым значением e 1E-5. Гены помечались как «неаннотированные», если либо 1) совпадение не было обнаружено в базе данных UniRef90, либо 2) совпадение было аннотировано следующими ключевыми словами: «неаннотированный», «неохарактеризованный», «гипотетический», «DUF» (домен неизвестен). функция).
Анализ белка McrA
Белок McrA, кодирующий геномы Methanogens и ANME, был выбран из списка идентификаторов доступа, найденного в приложении Shao et al. 35 . субконтиги, содержащие mcrA, экстрагировали не более чем с 15 генами до и после mcrA . Контекстуализированные и контекстуализированные внедрения McrA были рассчитаны с использованием ESM2 и gLM соответственно.
Распределения неаннотированных и аннотированных вложений
Распределения неаннотированных и аннотированных вложений в базе данных сравнивались с использованием дивергентного анализа Кульбака-Лейблера (KL). Сначала десять случайных выборок по 10 000 субконтигов из корпуса MGnify. Встраивания генов в pLM и gLM рассчитывали с использованием объединенных средних значений последнего скрытого слоя встраивания ESM2 и объединенных средних значений последнего скрытого слоя gLM соответственно. Выбросы были удалены с использованием расстояния Махаланобиса и порога хи-квадрат 0,975. Размеры встраивания pLM и gLM были сокращены до 256 главных компонентов (объяснены общие отклонения 91,9 ± 1,72% и 80,1 ± 6,89% соответственно). Дивергенцию KL рассчитывали по следующей формуле. ( 4 ).
$${D}_{{KL}}({P||Q})=frac{1}{2}left({{{{{ m{tr}}}}}}({Sigma }_{1}^{-1}{Sigma }_{0})-k+{({{mu }}_{1}-{{mu }}_{2})}^{T} {Sigma }_{1}^{-1}({{mu }}_{1}-{{mu }}_{0})+{{{{mathrm{ln}}}}} left(frac{det {Sigma }_{1}}{det 0} ight) ight)$$
(4)
где P соответствует распределению неаннотированных генов, а Q соответствует распределению аннотированных генов, где ({{mu }}_{0}, {{mu }}_{1}) соответственно в качестве средних значений и ({Sigma }_{0},{Sigma }_{1}) соответственно как ковариационные матрицы. Значимость различий в KL-дивергенции между встраиваниями pLM и gLM рассчитывается с использованием парного t-критерия для десяти образцов.
Прогнозирование числа ферментной комиссии
Пользовательский набор данных MGYP-Enzyme Commission (MGYP-EC) был создан путем первого поиска (mmseqs2 52 с настройкой по умолчанию) MGYP по набору данных «split30.csv», ранее использовавшемуся для обучения CLEAN 59 . Набор данных «split30.csv» состоит из номеров EC, присвоенных последовательностям UniProt, кластеризованным с идентичностью 30%. Учитывались только совпадения MGYP с последовательностями >70% для «split30.csv», а MGYP с несколькими совпадениями со сходством >70% были удалены. Тестовое разделение было выбрано путем случайного выбора 10% идентификаторов UniProt «split30.csv» в каждой категории EC, которые соответствуют MGYP. Категории EC с менее чем четырьмя различными идентификаторами UniProt ID с сопоставлением MGYP были удалены из набора данных, в результате чего было создано 253 категории EC. Набор поездов состоял из подконтигов MGnify в корпусе, который содержал по крайней мере один из 27936 MGYP, сопоставленных с 1878 идентификаторами UniProt. Тестовый набор состоял из случайно выбранного подконтига MGnify, содержащего каждый из 4441 MGYP, сопоставленного с 344 идентификаторами UniProt. Вложения pLM (бесконтекстные) рассчитывались для каждого из MGYP с присвоением номера EC путем объединения среднего значения последнего скрытого слоя его встраивания ESM2. Маскированные (только контекстные) внедрения gLM были рассчитаны для каждого из 19 слоев путем выполнения вывода по субконтигам с масками в позициях MGYP с присвоением номера EC и последующего извлечения скрытых представлений для каждого слоя для замаскированных позиций. Вложения gLM (контекстуализированные) рассчитывались также для каждого слоя путем выполнения вывода без маскировки и последующего извлечения скрытых представлений каждого слоя для MGYP с присвоением номеров EC. Для этих вложений было проведено линейное зондирование с одним линейным слоем. Линейные зонды обучались с ранней остановкой (терпение = 10, github.com/Bjarten/early-stopping-pytorch/blob/master/pytorchtools.py) и размером пакета = 5000, а результаты обучения повторялись пять раз со случайными начальными числами для расчета. диапазоны ошибок.
Дисперсия контекстуализированного анализа встраивания белка
Контекстуализированные встраивания белков генерируются во время вывода. Вариации контекстуализированных встраиваний белков были рассчитаны для MGYP, которые встречаются в наборе данных не менее 100 раз, исключая вхождения на краях субконтига (первый или последний токен). Для каждого такого MGYP мы берем 10 случайных независимых выборок, состоящих из 100 вхождений, и вычисляем средние попарные евклидовы расстояния между контекстуализированными вложениями. Чтобы оценить роль, которую gLM играет в контекстуализации, мы использовали описанный выше метод выборки для расчета дисперсии усредненных по контигам вложений pLM (среднее значение встраивания pLM, объединенных по всему контигу) для каждого MGYP, который встречается в наборе данных не менее 100 раз.
Анализ внимания
Головы внимания (n = 190) были извлечены путем вывода на немаскированных подконтигах, а необработанные веса внимания впоследствии были симметричны. E.coli K-12 RegulonDB 56 использовалась для исследования головок с моделями внимания, которые больше всего соответствуют оперонам. Корреляция Пирсона между симметричным первичным вниманием и оперонами была рассчитана для каждой головы. Мы обучили классификатор логистической регрессии, который предсказывает, принадлежат ли два соседних гена к одному и тому же оперону, на основе весов внимания во всех головках внимания, соответствующих паре генов.
Анализ структурных гомологов TnsC
Структурные гомологи TnsC были идентифицированы путем поиска ShCAST TnsC (цепь H PDB 7M99) в базе данных MGYP с использованием Foldseek 60 в ESM Atlas ( https://esmatlas.com/ ). Контиги, содержащие эти гомологи, в базе данных MGnify были использованы для расчета контекстуализированных встраиваний в белки идентифицированных структурных гомологов. Контиги с менее чем 15 генами были исключены из анализа. Контиги, кодирующие белки, которые ранее были идентифицированы как «TnsC» с использованием базы данных UniRef90 (см. раздел «Методы аннотации генов» выше), были включены в базу данных. «TnsC-подобные» контиги аннотировали вручную на основании наличия генов транспозазы (TnsB) и TniQ. Пятьдесят случайных примеров контигов MGnify, содержащих MGYP, аннотированных как NuoA и DnaB, были добавлены в качестве отрицательного контроля для визуализации UMAP. Мы рассчитали коэффициенты дивергенции KL, используя следующее уравнение. ( 5 ).
$$frac{{D}_{{KL}}(B{{{{{ m{||}}}}}}A)}{{D}_{{KL}}(C{{{ {{ m{||}}}}}}А)}$$
(5)
где A — распределение представлений известных TnsC, B — распределение представлений вручную курируемых TnsC-подобных регуляторов AAA+, C — распределение представлений других регуляторов AAA+, которые являются функционально несвязанными структурными гомологами известных TnsC. Следовательно, эта метрика находится в диапазоне от 0 до 1, где более низкое соотношение представляет собой повышенную способность функционально различать распределение B от C относительно A. Дивергенция KL рассчитывалась по той же формуле, что и в разделе «Методы». Распределение неаннотированных и аннотированных вложений, за исключением с 20 главными компонентами, которые объясняли >85% дисперсий во всех вложениях.
Анализ паралогии и ортологии
Идентификаторы UniProt из пар паралогов ABC-транспортера ModA и белка ModC ( n = 4823) были идентифицированы ранее Овчинниковым с соавт. 47 и были загружены с https://gremlin.bakerlab.org/cplx.php?uni_a=2ONK_A&uni_b=2ONK_C и впоследствии использовались для загрузки необработанных последовательностей белка с сервера UniProt. Для последующего анализа были выбраны только пары ( n = 2700), где обе необработанные последовательности были доступны для загрузки и где идентификатор UniProt отличался на единицу (что указывает на соседнее положение в эталонном геноме). Мы построили тестовые контиги, состоящие из трех генов, где первый и третий гены замаскированы, а второй ген кодирует один из пары в прямом направлении. Затем мы запросили gLM, чтобы предсказать два соседних замаскированных гена, и считали предсказание правильным, если любой из белков, наиболее близких к предсказанию замаскированных генов с наибольшей достоверностью в пространстве встраивания, принадлежит к тому же кластеру последовательностей, что и взаимодействующий белок (50% аминокислотной последовательности). идентичность, рассчитанная с использованием CD-HIT v4.6 61 ). Случайная вероятность правильного прогнозирования (1,6 ± 1,0) была смоделирована с использованием 1000 итераций случайных прогнозов, сгенерированных в рамках стандартного нормального распределения, и выполнения той же операции, что и выше, для вычисления уровня правильных прогнозов 62 .
Таксономический анализ и визуализация
12 февраля 2023 года из базы данных RefSeq ( ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq ) был загружен 4551 репрезентативный геном бактерий и архей и 11660 эталонных вирусных геномов. Выбирается случайный подконтиг из 30 генов и закодированные с использованием ESM2, которые затем были объединены с вектором ориентации и затем использованы в качестве входных данных для обученного gLM. Последний скрытый слой был объединен по средним значениям по всей последовательности для получения контекстуализированных вложений контигов из 1280 объектов. Встраивания белка ESM2 также были объединены по средним значениям по всей последовательности для извлечения 1280-функциональных внеконтекстных встраиваний контигов. Мы обучили классификатор логистической регрессии прогнозировать таксономию подконтигов на уровне класса и оценили производительность с помощью стратифицированной k-кратной перекрестной проверки (k = 5).
UMAP-визуализация и статистические тесты
Все уменьшения размерности UMAP рассчитаны со следующими параметрами: n_neighbors = 15, min_dist = 0,1. Оценки силуэта были рассчитаны с использованием пакета sklearn с использованием настроек по умолчанию и метрики евклидового расстояния.
Сводная отчетность
Дополнительную информацию о дизайне исследования можно найти в сводке отчетов о портфеле природы, связанной с этой статьей.
Доступность данных
Набор данных, используемый для обучения, доступен для загрузки с сервера MGnify ( http://ftp.ebi.ac.uk/pub/databases/metagenomics/peptide_database/2022_05/ ). Модель доступна на zenodo под номером 10.5281/zenodo.7855545. Исходные данные для основного рисунка ( 2c ,e,f,i&h; 3a,b,c&d ; 4a,b,c,e&f ; 5b,c,d,e&f ) и дополнительных рисунков . ( 2 , 3 , 4 , 5 , 7 , 8 , 9 , 10 и 11 ) прилагаются к этому документу в виде zip-файла. Исходные данные приведены в статье.
Источник: pcr.news