Чем заняться в биоинформатике
МЕНЮ
Главная страница
Поиск
Регистрация на сайте
Помощь проекту
Архив новостей
ТЕМЫ
Новости ИИ
Городские сумасшедшие
ИИ в медицине
ИИ проекты
Искусственные нейросети
Искусственный интеллект
Слежка за людьми
Угроза ИИ
ИИ теория
Компьютерные науки
Машинное обуч. (Ошибки)
Машинное обучение
Машинный перевод
Нейронные сети начинающим
Психология ИИ
Реализация ИИ
Реализация нейросетей
Создание беспилотных авто
Трезво про ИИ
Философия ИИ
Генетические алгоритмы
Капсульные нейросети
Основы нейронных сетей
Промпты. Генеративные запросы
Распознавание лиц
Распознавание образов
Распознавание речи
Творчество ИИ
Техническое зрение
Чат-боты
Авторизация
2020-05-17 15:00
Вообще, принципы автоматизации, анализа данных пришли во многие естественные науки - возникла хемоинформатика, геоинформатика (например анализ геодезии или модели метеорологических предсказаний, основанных на большом количестве параметров), астрофизика, которая обсчитывает данные с многих телескопов и пытается на их основе объяснять физические явления.
В биологии сошлось много факторов: данные с самого начала стали собирать в единые базы данных, в едином формате. Сами данные (обычно это последовательности букв) состоят из большого количества элементов - от сотен до миллиардов, что дает результатам обработки хорошую статистическую поддержку и высокое разрешение. Появилось много относительно недорогих методов получения этих данных. Большинство задач на анализ последовательностей относительно просто решать с алгоритмической точки зрения - это вам не считать векторы того, куда дует какой ветер или как движутся галактики. И, в конце концов, высочайший спрос на рынке, в связи с заинтересованностью людей в медицине. Возможно, описанные в этой статье методы когда-то станут неактуальными, но общие принципы останутся неизменными. Итак, разберемся в том, чем же можно заняться в современной биоинформатике. Правда, затронуты будут не все, а лишь самые известные методы. О теоретических основах, методах выравнивания последовательностей, секвенировании и эволюционной геномике можно прочитать в нашем пособии. В этой статье я подробно останавливаться на этом не буду.
В открытом для всего человечества доступе находятся терабайты информации в биологических базах данных. На них одних можно даже делать какие-то исследования. Но в основном биоинформатик работает с данными, полученными в результате эксперимента. Например, группа «мокрых» (экспериментальных) биологов подготовила культуру клеток в специальных условиях и культуру клеток без внешних воздействий. Потом они извлекли из двух культур какие-то данные. Обычно это результаты секвенирования ДНК. Про секвенирование по Сэнгеру я уже писал, но сейчас мало кто им пользуется на практике. Большую популярность приобрели более дешевые и производительных технологии секвенирования нового поколения (NGS - Next Generation Sequencing). Известны такие бренды как Illumina и Oxford Nanopore, но есть и много других.
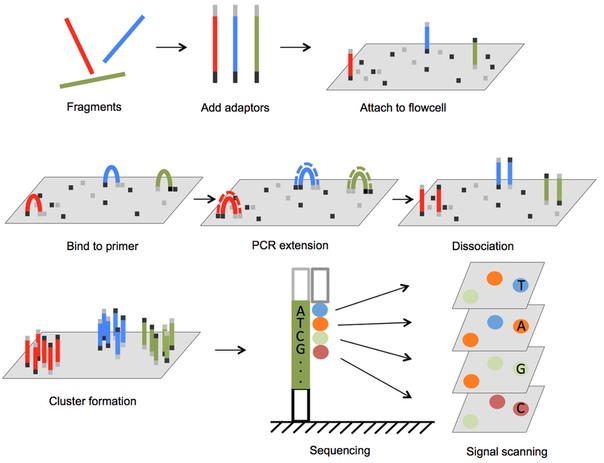
Так или иначе, они выдают файлы длины ~150 пар оснований в случае Illumina и до ~10 тысяч пар оснований в случае Nanopore в формате FastQ (Fasta with quality). Выглядит он как последовательность нуклеотидов, к которой добавлена мера качества прочтения (в виде странных символов строкой ниже самих букв):
FastQ-формат:
@Seq_ID ATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCC + ''*((((***+))%%%++5(%%%%>.1*CF-+*''))**
Далее, с помощью особых методов, проводится контроль качества и дальнейшая очистка. В результате мы можем получить, например, файл в формате Fasta:
Fasta-формат:
>Seq_ID ATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCC
Поскольку, в силу особенностей методов секвенирования, мы получили лишь фрагменты генома (если мы конечно секвенировали геном), нам нужно собрать его в целый. Затем хорошо бы определить, например, что к каким хромосомам относится, аннотировать геном (то есть сопоставить закодированным белкам или РНК). Сборка генома - задача не из простых:
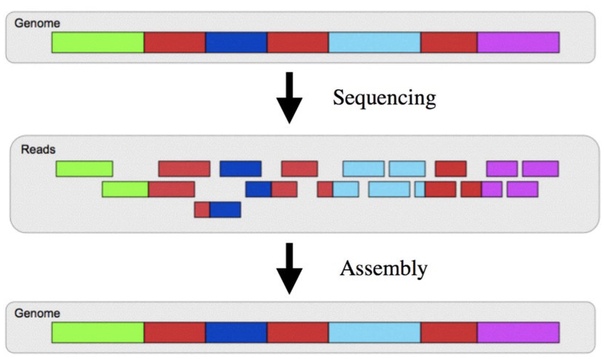
Чтения по Сэнгеру были длинными и качественными, но дорогими. В те времена геномы собирали с помощью алгоритмов поиска перекрытия последовательностей.
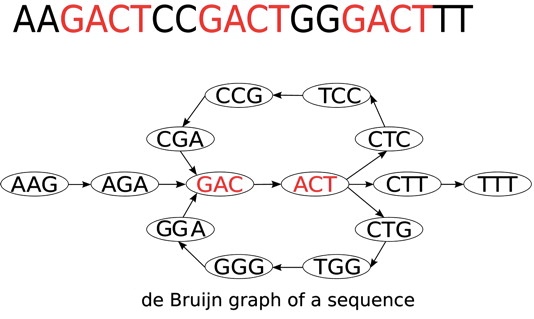
NGS-данные очень дешевые (потому популярные), но длина прочтения стала ниже, а качество упало. Поэтому в этом случае задачу решают с помощью графов де Брюйна.
Итак, мы собрали геном, или даже воспользовались уже собранным геномом. Теперь нам доступны различные виды анализа
— Мы можем выровнять последовательности и построить филогенетическое дерево (на самом деле это редко делается на полных геномах). Этому вопросу была посвящена методичка.
— Можно рассмотреть несколько геномов очень близких видов/подвидов/популяций, изучить в них SNP (single nucleotide polimorphisms — однонуклеотидные полиморфизмы) для изучения точечных мутаций, хромосомные перестройки и другие эволюционные события. Особенно актуально в плане изучения человеческих популяций, представителей разных рас и народностей.
— Можно провести эксперимент, например мы хотим узнать, что произойдет с геномом в результате облучения радиаций. Для этого мы должны облучить ее и отсеквенировать. В клетках произойдут мутации и перестройки, а короткие прочтения секвенатора позволят картировать это все на исходный геном, чтобы выяснить, где эти мутации происходят и насколько они случайны.
Можно модифицировать наш эксперимент и изучать не только DNA, но и RNA, и даже DNA-белковые взаимодействия, более того, не очень большая длина прочтений нам в этом поможет.
ChIP-seq
Если растянуть всю DNA человека в одну линию, ее длина составит 2 метра. На самом деле, в живых организмах она плотно упакована. У эукариот первым уровнем компактизации являются нуклеосомы. Они состоят из нескольких белков гистонов, на которые намотана DNA определенной длины. В общем, транскрипция у эукариот в основном регулируется за счет изменения компактизации DNA и формирования эухроматина/гетерохроматина, поэтому данные о модификации гистонов или о посадке транскрипционных факторов на конкретных участках DNA могли бы многое сказать об экспрессии. Этим и занимается метод секвенирования с иммунопреципитацией хроматина (chromatin immunoprecipitation, ChIP).
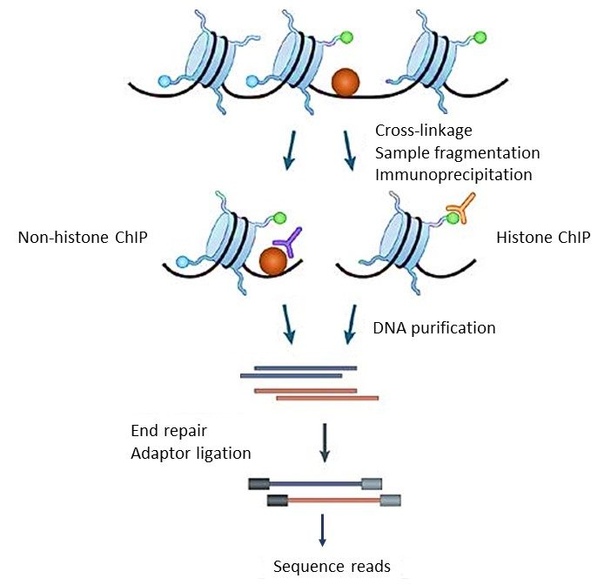
Сначала DNA «склеивают» с белками формальдегидом, потом разрезают ультразвуком или эндонуклеазами по «мостикам» между DNA-белковыми комплексами. Затем осаждают специфическими к исследуемому белку антителами и очищают DNA от белков. Затем секвенируют. Полученные фрагменты картируют на геном и выясняют сайты связывания белков с DNA. Сейчас разрабатываются методы, которые позволят выяснить взаимодействие вообще всех белков с DNA, а не только гистонов.
Hi-C и TAD
Как я уже говорил, DNA в ядре эукариот плотно упакована. А что, если мы хотим выяснить не модификацию гистонов, а что-то по-крупнее? Семейство таких методов называют определением пространственной конформации хромосом (chromosome conformation capture, 3C). В результате мы получаем карту топологически-связанных доменов (topologically associated domains), чтобы например выяснить взаимодействие между промотором и энхансером. Регуляторные элементы могут располагаться на расстоянии нескольких миллионов пар оснований от генов, которые они контролируют, именно поэтому этот метод интересен. Раньше микроскопия была основным методом исследования ядерной организации, сейчас ее данные совмещают с Hi-C.
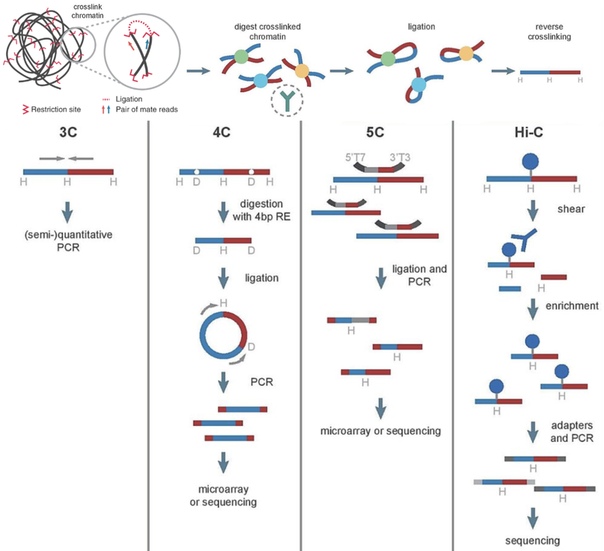
Процедура метода похожа на процедуру ChIP-seq. Сначала склеивают белки с DNA формальдегидом, затем разрезают. Но потом производят лигирование разрезанных участков, чтобы рядом находящиеся фрагменты оказались в склеены. Затем DNA чистят и подвергают другим операциям, в зависимости от метода. Затем секвенируют. Если картировать риды на геном и обработать, можно получить тепловую карту контактов, в которой более темными треугольничками будет показаны ассоциированные участки генома. Квадраты получаются в результате симметрического отражения таких треугольников.
RNA-seq
В геноме закодированы гены. С DNA считывается RNA, а с неё — белок. В одном организме все клетки имеют одинаковый геном (в некотором приближении). Функциональные различия между клетками разных тканей создаются не за счет того, что гены разные, а за счет того, что работают они по-разному. Также, клетки могут различаться, находясь в разных условиях. Степень работы геном можно оценить мерой экспрессии. Для определения экспрессии генов существует множество «мокрых методов», таких как гель-электрофорез, нозерн-блот (если мы исследуем RNA), вестерн-блот (если мы исследуем белки), количественная ПЦР, микрочипирование и другие. Сейчас нам более интересно рассмотреть метод секвенирования RNA.
Вообще, метод позволяет изучать весь траскриптом, то есть не только матричные RNA, но и разнообразные некодирующие RNA. Пока что мы умеем секвенировать только DNA, поскольку для этого процесса существуют термостабильные DNA-зависимые-DNA-полимеразы, а для ПЦР нужны RNA-праймеры. Поэтому, выделив из образца всю RNA, мы должны провести обратную транскрипцию, которую подсмотрели у ВИЧ. В итоге мы секвенируем кДНК (комплементарную ДНК). Полученные сиквенсы часто идентичны отдельным молекулам RNA. Потом мы картируем их на геном и занимаемся анализом. RNA-секвенирование позволяет получать данные о вариантах сплайсинга, пост- и ко-трансляционном редактировании РНК и SNP. Кроме того, РНК-секвенирование позволяет получить абсолютную количественную информацию о представленности различных транскриптов в пробе, в отличие от относительных количественных данных вышеперечисленных мокрых методах. Совершенствование технологий секвенирования РНК наряду с развитием секвенирования РНК одиночных клеток (single-cell RNA-seq) позволяет более детально изучать этиологию и патогенез различных заболеваний.
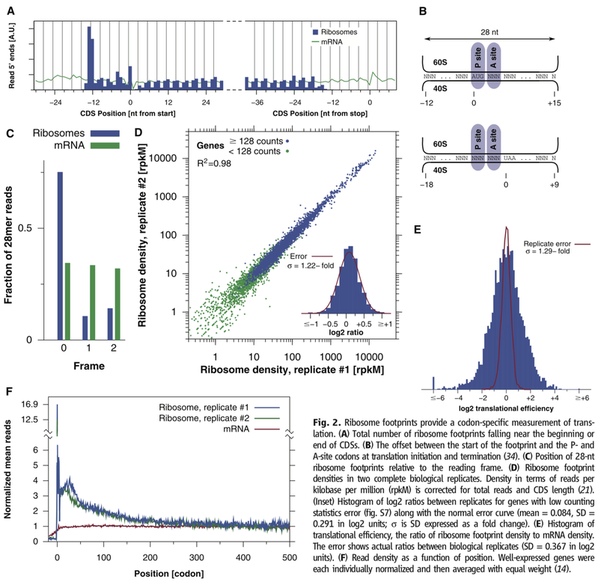
Рибосомный профайлинг
Равно как на DNA есть регуляторные элементы, ответственные за транскрипцию, так и на полученной mRNA есть элементы, регулирующие трансляцию. Все они по большей части управляют посадкой рибосомы и рибосомальных факторов на молекулу RNA. А что, если мы как бы запечатлеем перемещение рибосомы при трансляции «в моменте»? Примерно этим занимается метод Ribo-seq. На самом деле, это модифицированный RNA-seq, только читаем мы не всю RNA. Мы массово секвенируем фрагменты mRNA, взаимодействующие с рибосомами. Этот метод позволяет наблюдать за трансляцией, конкуренцией рибосом за локализацию вокруг mRNA в определенном месте. Также его используют для нахождения области, кодирующей белки в mRNA, рамок считывания, для определения месторасположения рибосом на mRNA. К сожалению, метод не позволяет прямо описывать кинетику элонгации и остановки рибосом, задействованных в процессе элонгации. Таким образом, мы можем следить за трансляцией, которая не всегда происходит с AUG-кодона и следить за скоростью перемещения рибосом, а следовательно и параметрами фолдинга белка.
Схема эксперимента простая: фиксируют рибосомы на RNA с помощью циклогексимида, разрушают RNA, не связанную с рибосомами при помощи рибонуклеаз, выделяют rRNA-рибосомные комплексы методом ультрацентрифугирования в градиенте сахарозы, чистят, а дальше все как в RNA-seq. В итоге получаются только кусочки, находившиеся внутри рибосом.
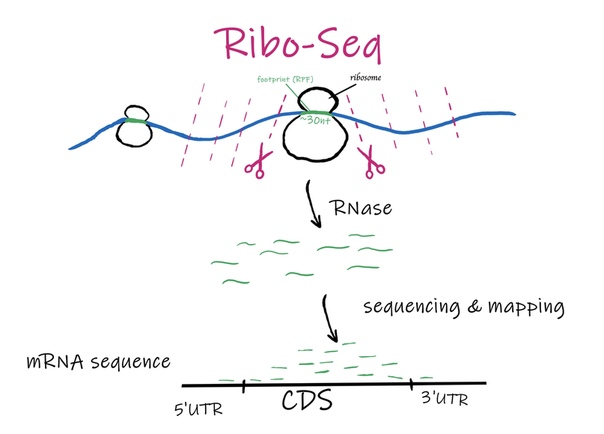
В результате анализа получаются статистические данные распределения рибосом на RNA, рамки считывания и другое.
Моделирование биомолекул
Особняком стоит исследование структуры биополимеров. В российской практике это называется структурной биоинформатикой, а за рубежом — разделом биохимии или хемоинформатики. В любом случае, чтобы этим заниматься, нужно получить биологическое образование, поэтому я и включил этот раздел в статью.
Данные для анализа добываются следующим образом: из клеток выделяют и кристаллизуют белки. Звучит просто, но на деле для каждого белка приходится придумывать индивидуальный способ кристаллизации. Затем с кристаллом проводят рентген-структурный анализ. В итоге через анализ электронной плотности, получают файл с координатами атомов в формате PDB. Существуют специальные визуализаторы, основанные на языках программирования Java (JMol), Python (PyMol), Chimera и другие.
Есть интересный программный пакет PyRosetta (почти как Розеттский камень), который позволяет генерировать структуру молекул по заданной последовательности и программа VinaAutoDock, позволяющая осуществлять молекулярный докинг (предсказывать место возможного сайта связывания с молекулой) — так сейчас дизайнят лекарства. А еще есть молекулярная динамика — способ изучения того, как меняется конформация молекул во время фолдинга или при взаимодействии с лигандом.
Машинное обучение
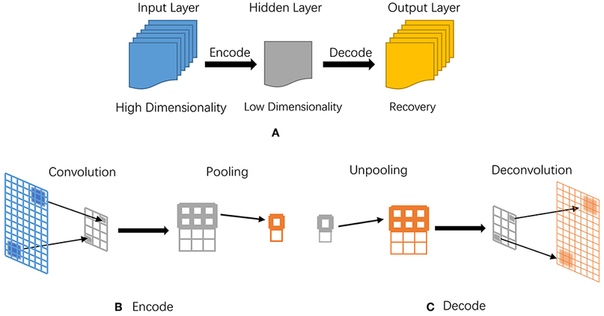
В последнее время в биоинформатику проникает Big Data анализ и машинное обучение, то есть написание нейросетей. Нейросеть — это даже не алгоритм, а некоторая математическая модель, воплощенная в виде программного кода, которая осуществляет обучение на большой выборке, и научившись, она может самостоятельно что-то узнавать или предсказывать. Все этим сейчас занимаются — чем же биология хуже? В ней есть огромные базы данных, есть спрос на предсказание и узнавание. Каждая задача, решаемая таким способом уникальна, поэтому нет смысла их рассматривать в рамках статьи. Для понимания: можно скормить нейросети транскриптом бактерии в паре тысяч разных условий и попросить ее предсказать профиль экспрессии для каких-то новых условий. Нейросеть типа autoencoder сжимает данные до заданного минимума, а потом разворачивает. Таким образом, данные проходят через бутылочное горлышко, и так иногда удается найти важные для системы элементы. Нейросеть-предиктор позволяет предсказывать скрытые или неизвестные значения на основании имеющихся данных, в основном, по другим признакам. Сеть-дискриминатор определяет, насколько полученные данные соответствуют действительности.
Python, R и командная строка
Я все время упоминал «написать алгоритм» или «статистически обработать». Что это все вообще значит? А значит это, что, раз мы имеем дело с компьютером, нужно научиться программировать. Имеется, в виду, не нужно быть гением во frontend или backend разработке сайтов. Многие алгоритмы уже написаны и существуют в виде пакетов, нужно лишь научиться их грамотно применять, в зависимости от ситуации. Все-таки, биоинформатик —это в первую очередь биолог, который должен обладать биологическим чутьем и понимать основы основ жизни. В этой связи, упомяну, в чем именно работают биоинформатики.
Командная строка и Bash. Она по умолчанию установлена на Unix операционных системах, наиболее интересными являются Linux и Mac, в связи с чем, рекомендую их использование разного рода биоинформатикам. В ней очень удобна работа с файлами и директориями, удаленное управление вычислительным сервером и обработка файлов. Ну, например, вам нужно заменить в текстовом файле, весом десятки гигабайт, обозначение хромосомной локализации «chr4» на «chr_4». Тот же ворд умрет от такого размера файла, да и не все умеет делать автоматически. Еще в интерфейсе командной строки можно поместить множество различных алгоритмов, написанных на разных языках программирования.
Python. Не очень сложный в плане синтаксиса язык используется, грубо говоря, как продвинутый «калькулятор», который умеет применять различные функции, циклы, работать с файлами, в нем пишут разнообразные скрипты. Для справки, если вы хотите разработать графический интерфейс приложения, лучше выучить язык С++ или С#. В питон можно подгружать программные пакеты, многие из которых специально написаны для биологов.
R. Модный нынче язык для работы со статистикой. Грубо говоря, являет собой сложный Excel, который позволяет вычислять матожидание, медиану, устраивать проверку по статистическим критериям и, самое крутое, рисовать графики (к слову, для этого питон тоже годится)!

Получается, для занятия биоинформатикой, вам всего лишь нужен секвенатор, и если быть точнее, секвенирование проведут за вас мокрые биологи, а вам вышлют данные для анализа. В зависимости от постановки эксперимента, мы можем получить с одного секвенатора самые разнообразные биологические данные. Все, что нужно для работы — это компьютер и сервер для вычислений. Вот такой красивой может быть наука, столько интересного можно извлечь из, казалось бы простых последовательностей, длиной в 150 букв.
Телеграм: t.me/ainewsline
Источник: m.vk.com