Синдром Лея
МЕНЮ
Главная страница
Поиск
Регистрация на сайте
Помощь проекту
Архив новостей
ТЕМЫ
Новости ИИ
Городские сумасшедшие
ИИ в медицине
ИИ проекты
Искусственные нейросети
Искусственный интеллект
Слежка за людьми
Угроза ИИ
ИИ теория
Компьютерные науки
Машинное обуч. (Ошибки)
Машинное обучение
Машинный перевод
Нейронные сети начинающим
Психология ИИ
Реализация ИИ
Реализация нейросетей
Создание беспилотных авто
Трезво про ИИ
Философия ИИ
Генетические алгоритмы
Капсульные нейросети
Основы нейронных сетей
Промпты. Генеративные запросы
Распознавание лиц
Распознавание образов
Распознавание речи
Творчество ИИ
Техническое зрение
Чат-боты
Авторизация
2020-03-24 18:10

Синдром Лея (СЛ) — это гетерогенное генетически обусловленное заболевание, относящееся к группе митохондриальных энцефаломиопатий с аутосомно-рецессивным или митохондриальным типом наследования и связанное с мутациями в генах, кодирующих полипептиды комплексов дыхательной цепи митохондрий, а также белков, принимающих участие в их сборке на внутренней поверхности митохондриальной мембраны [4].
Первые описания данного синдрома были даны Денисом Леем в 1951 году [1].
На заре открытия данного заболевания появились предположения, что его причиной является нарушение метаболизма, однако они не получили своего клинического подтверждения.
Доктор F. Hommes в 1968 году описал семьи, у представителей которых наблюдалось снижение активности пируваткарбоксилазы [2]. С 1975 года появились данные, что причиной СЛ может являться недостаточность пируватдегидрогеназного комплекса, а позже были обнаружены молекулярно-генетические нарушения. С развитием генетики и биохимии было выяснено, что изменения генов в мтДНК, которые ответственны за кодирование субъединицы АТФ-азы или тРНК — ядерных генов, кодирующих полипептиды комплекса дыхательной цепи митохондрий, — а также за нарушения в генах, отвечающих за сборку КДЦМ на митохондриальной мембране, приводят к развитию синдрома Лея [3]. Всего насчитывается 12 генов: NDUFS4, NDUFS5, NDUFS6, NDUFS7, NDUFS8, NDUFV1, SDHA, SURF1, COX10, COX15, SCO2, BCS1L [1].
Чаще всего дефект обнаруживается в результате недостаточности IV КДЦМ-цитохром С-оксидазе (COX). Этот фермент является последним в электронно-транспортной системе митохондрий. СОХ включает в себя 13 субъединиц: 11 кодируются ядерными генами, 3 – мтДНК.
Чаще всего данная патология возникает в результате мутации гена SURF1, который лежит в хромосоме 9q34, кластере 6. Данный белок включен во внутреннюю мембрану митохондрии, и нарушения в нем приводят к синтезу укороченного белка и, как следствие, к повреждению СОХ-комплекса [4].
В результате биохимических исследований у большинства пациентов с СЛ обнаруживается повышение лактата в крови и спинномозговой жидкости. Повышение соотношения лактат/пируват является отражением нарушения окислительно-восстановительного баланса в цитоплазме [4].
Те пациенты, у которых мутации не превышают 70% от всех мтДНК к конкретной ткани, не имеют клинических проявлений данного синдрома. Однако при превышении данного порога могут проявляться симптомы, которые будут описаны ниже [1].
Синдром Лея чаще всего проявляется в детстве в виде утраты уже имеющихся психомоторных навыков, мозжечковыми и экстрапирамидными расстройствами, судорогами, мышечной гипотонией. Однако могут проявляться и такие симптомы как нистагм, потеря слуха, атрофия зрительного нерва. Дети отстают в своем развитии, отмечается регресс психомоторных функций, вялость, сонливость. Такие пациенты склонны к лактоацидозам. Ночью и при физических нагрузках возникают проблемы с дыханием в виде апноэ, диспноэ, тахипноэ [1].
При МРТ-диагностике наблюдается снижение МР-сигнала в различных областях ГМ.
Также пациентам с синдромом Лея свойственна «парадоксальная гиперкетонемия» — повышение уровня кетоновых тел после пищевой нагрузки и высокое соотношение 3-гидроксибутират/ацетоацетат в крови. При проведении анализа органических кислот мочи может наблюдаться повышенная экскреция органических кислот, участвующих в цикле Кребса (фумаровая, янтарная и др.) [4].
Синдром Лея имеет следующую классификацию:
- типичный СЛ без эпилепсии и кардиомиопатии, обусловленный дефектом гена SURF1 (локус 9q34);
- Лей-подобный синдром, обусловленный мутацией гена SCO2, локус 22q13;
- СЛ с дистонией, тугоухостью и метилмалоновой ацидурией (дефект гена SUCLA2, локус 13q12.2-q13);
- СЛ с нефрозом или почечной недостаточностью (дефект биосинтеза субъединицы CoQ1, ген PDSS2, локус 6q21);
- Лей-подобный синдром с флюктуирующей периферической нейропатией и дистонией с лактатацидозом (дефект гена PDHA1, локус Xp22.2-p22.1);
- ювенильная форма СЛ с атрофией зрительных нервов (мутация гена TACO1, локус 17q22-q24.2) [1].
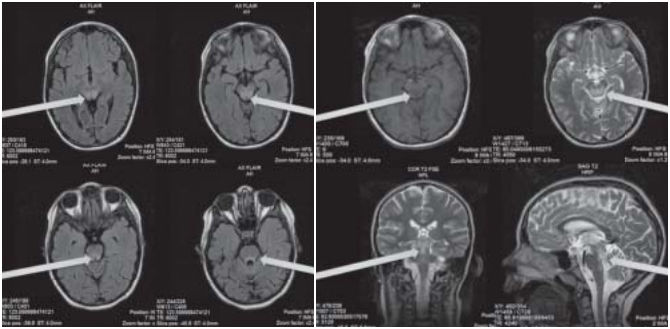
Рисунок 1
На представленных МРТ-изображениях обращают на себя внимание распространенные зоны повышения МР-сигнала на Т2 ВИ и во FLAIR по задней поверхности ствола. Желудочковая система незначительно вторично расширена, ликвородинамика компенсирована. Перивентрикулярные зоны интактны. Гиппокампальные и парагиппокампальные регионы не изменены. Гипоталамо-гипофизарный регион без очаговых нарушений МР-сигнала. Краниовертебральный переход сформирован правильно [1].
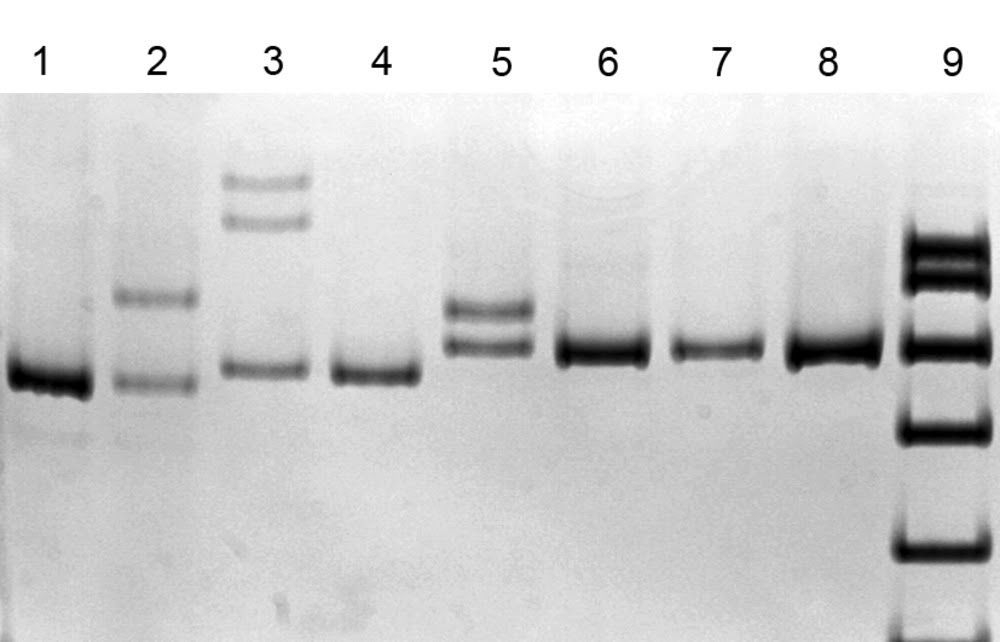
ДНК-диагностика 3 частых мутаций в гене SURF1 с помощью метода SSCP анализа (электрофорез в 8% полиакриламидном геле) [4]..
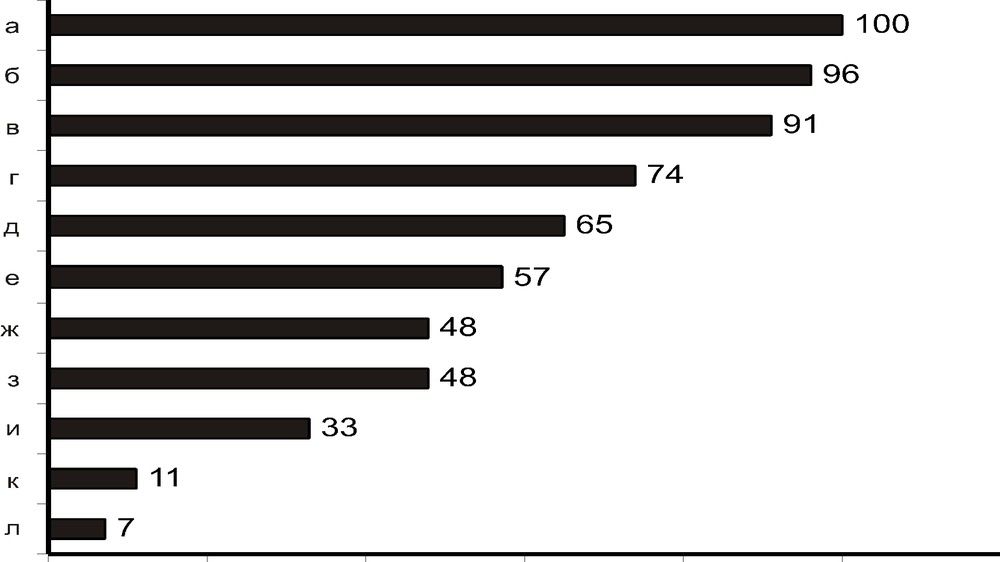
Статистика основных синдромов [4]:
- а — задержка психомоторного развития;
- б — мышечная гипотония;
- в — гипертрихоз;
- г — мозжечковый синдром;
- д — офтальмопарез;
- е — полинейропатический синдром;
- ж — дыхательные нарушения;
- з — пирамидный синдром;
- и — частичная атрофия зрительных нервов;
- к — экстрапирамидные расстройства;
- л — эпилептический синдром.
Источники:
- И. Г. Ковалев. Синдром Ли (подострая некротизирующая энцефаломиелопатия). Описание клинического случая. / И. Г. Ковалев, А. А. Соломасова, В. А. Чадаев, Э. Ю. Волкова, А. А. Холин, Н. Н. Заваденко // Вестник РГМУ. Неврология. — 2012 №2 — С. 31–35.
- Van Der Knaap M. S. / Valk J. Magnetic Resonance of Myelination and Myelin Disorders. // Berlin Springer Verlag — 2005 — С. 1084–1085.
- Hommes F., Leigh's encephalomyelopathy: an inborn error of gluconeogenesis. / Polman H., Reerink J // Arch Dis Child — 2008 — C. 423–426.
- П. Г. Цыганкова. Синдром Ли, обусловленный мутациями в гене SURF1: клинические и молекулярно-генетические особенности. / П. Г. Цыганкова, С. В. Михайлова, Н. А. Пичкур // Журнал неврологии и психиатрии им. С. С. Корсакова — 2010 — С. 25–32.
Телеграм: t.me/ainewsline
Источник: medach.pro