Протокол эпидемии: что может сказать биоинформатика о вспышке nCoV
МЕНЮ
Главная страница
Поиск
Регистрация на сайте
Помощь проекту
Архив новостей
ТЕМЫ
Новости ИИ
Городские сумасшедшие
ИИ в медицине
ИИ проекты
Искусственные нейросети
Искусственный интеллект
Слежка за людьми
Угроза ИИ
ИИ теория
Компьютерные науки
Машинное обуч. (Ошибки)
Машинное обучение
Машинный перевод
Нейронные сети начинающим
Психология ИИ
Реализация ИИ
Реализация нейросетей
Создание беспилотных авто
Трезво про ИИ
Философия ИИ
Генетические алгоритмы
Капсульные нейросети
Основы нейронных сетей
Промпты. Генеративные запросы
Распознавание лиц
Распознавание образов
Распознавание речи
Творчество ИИ
Техническое зрение
Чат-боты
Авторизация
2020-02-15 23:22

Мы находимся в разгаре эпидемии новой опасной инфекции, но достоверной информации о ней мало. В этой ситуации помогает молекулярная эпидемиология. Ход накопления мутаций эволюционирующим вирусом позволяет оценить важные параметры эпидемии даже при неточных официальных данных. Ситуация очень быстро меняется; все оценки, приведенные ниже, верны по состоянию на 9 февраля.
С чего все началось?
Сравнивая организмы друг с другом, эволюционные биологи многое могут узнать про их историю. Больше всего материала для сравнений содержится в нуклеотидных последовательностях, в случае коронавирусов — в последовательностях РНК. Текущая вспышка — пожалуй, первый случай возникновения нового патогена, когда генетические данные есть почти с самого начала. Первая последовательность «нового» коронавируса (nCoV) появилась в открытом доступе всего через несколько недель после описания первых случаев [1]; для сравнения: при вспышке SARS (тяжелого острого респираторного синдрома, также вызываемого коронавирусами) 2002/2003 года первых последовательностей пришлось ждать много месяцев.
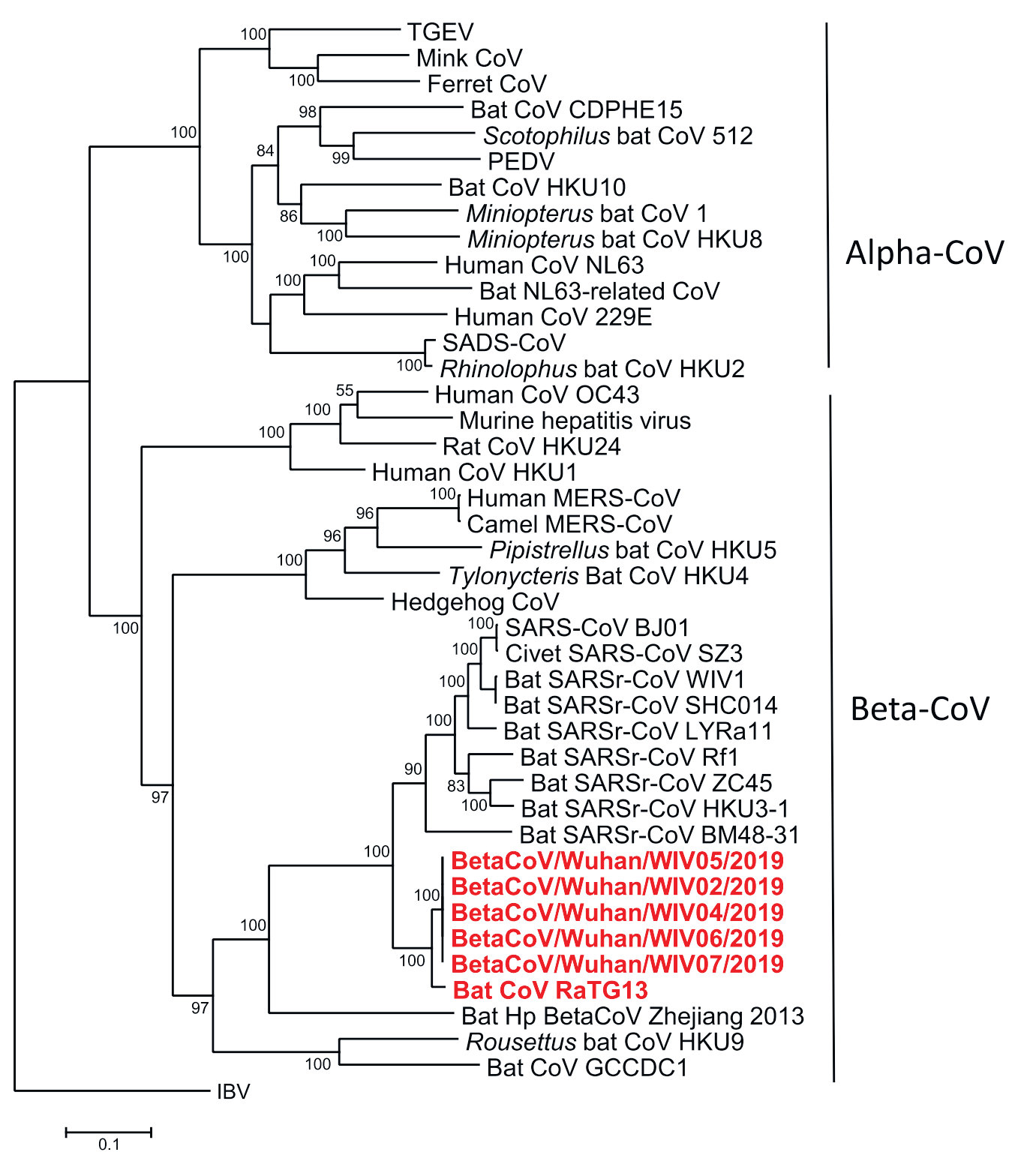
На рис. 1 показано эволюционное дерево коронавирусов. Как и на обычных эволюционных деревьях, расстояние между любыми двумя ветвями здесь соответствует времени, прошедшему с момента расхождения двух линий от их общего предка (LCA — last common ancestor). Видно, что ближайший родственник группы вирусных штаммов, давших начало этой вспышке, — это коронавирус, выделенный из летучей мыши; между ним и эпидемическими штаммами совпадает около 96% нуклеотидов. Такая картина может означать, что вирус был передан от летучих мышей, хотя окончательные выводы о пути передачи делать рано; в случае SARS, чтобы разобраться, потребовались годы. (Сведения о еще более близких штаммах из панголина пока есть только в виде пресс-релиза [2]). В любом случае все имеющиеся данные свидетельствуют о том, что инфекция была получена из природного резервуара. Чуть дальше расположены штаммы SARS, с которыми совпадает ~80% нуклеотидов; а еще дальше — штаммы MERS, ближневосточного респираторного синдрома [3, 4]. В последовательностях nCoV нет никаких свидетельств искусственной рекомбинации, вставок необычных фрагментов или какого-либо вмешательства; все сообщения об обратном, появлявшиеся в последние дни на препринтных серверах, были отозваны и/или опровергнуты.
Все линии nCoV более близки друг к другу, чем к любой другой известной вирусной последовательности. Это, по-видимому, означает, что занос был единственным. Не всегда бывает так: например, вспышки MERS в разные годы вызывались новыми переносами из природного резервуара — верблюдов [5].
Зная скорость эволюции (см. ниже), можно датировать LCA. По-видимому, он существовал в конце ноября — начала декабря. LCA мог быть у единственного человека, который заразил потом других, или же у животного, от которого заразилось несколько человек — это установить трудно. Первые описанные случаи датируются началом декабря; это означает, что вспышка была обнаружена почти сразу. Образцы вируса, выделенные с рынка в Ухане, очень близки к таковым у самых ранних пациентов из Уханя; это подтверждает, что первые люди заразились именно там.
Неизвестно, как мутации, отличающие nCoV от штаммов, распространенных у животных, изменили его характеристики и изменили ли вообще. Возможно, перескок был несчастной случайностью, не сопровождавшейся никакими изменениями генетических характеристик вируса по сравнению с его предками у диких животных.
Что происходит сейчас?
Любые биологические объекты изменяются в ряду поколений из-за случайных мутаций. «Перескочив» в людей, вирус продолжил эволюционировать, «прорастив» ветвистое эволюционное древо уже в людях (рис. 2) К сожалению, сегодня известны лишь очень немногие «листья» этого древа. Самые новые последовательности из Уханя датируются 3 января, и 42 из 73 известных сегодня последовательностей получены снаружи Китая (при том, что 99% подтвержденных случаев — в Китае [6]). Тем не менее, изучая древо, можно понять довольно много.
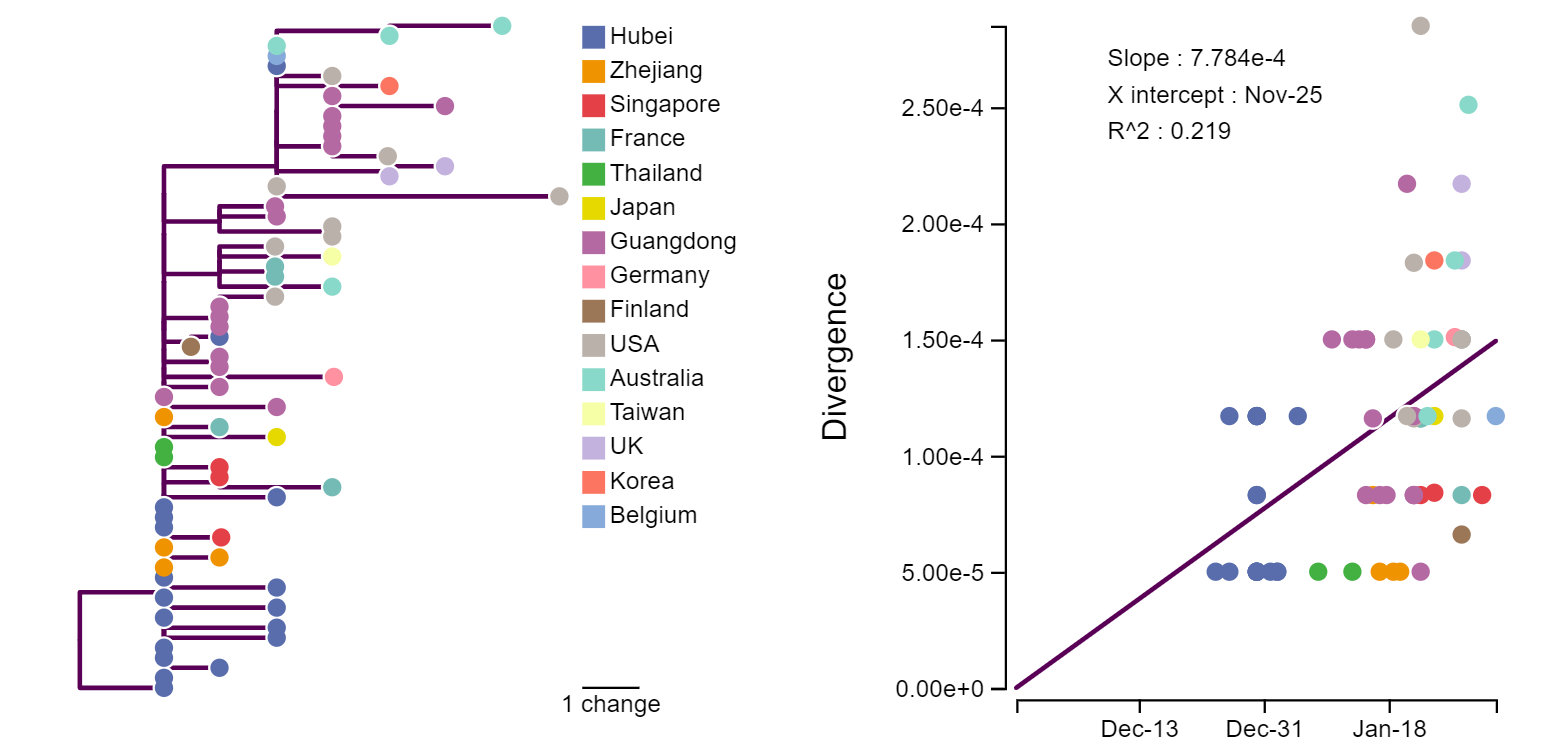
Во-первых, можно установить скорость эволюции вируса. Для этого нужно сопоставить даты инфицирования с числом отличий последовательности вируса от «предковой». По текущим оценкам, скорость составляет порядка 10–3 замен на нуклеотид в год [7, 8]; это сопоставимо с таковой у других РНК-содержащих вирусов, например, у гриппа [9]. Это может означать, что, как и в случае с гриппом, трудно будет создать универсальную вакцину, защищающую от всех штаммов, и вакцина потребует периодического обновления.
Во-вторых, можно проследить пути передачи. Первые надежные данные о передаче от человека человеку были получены именно таким образом. Можно задавать и более тонкие вопросы. Сколько людей заражается от членов семьи, сколько на работе, сколько в транспорте, сколько в больнице? Насколько эффективны карантинные меры? Как именно вирус переносится между странами? Можно пытаться узнать это по молекулярным данным. Это очень помогает с другими инфекциями, для которых таких данных больше, например с ВИЧ [10].
В-третьих, можно оценить скорость распространения вируса, а именно ставший знаменитым в последние дни показатель R0. R0, или базовое репродуктивное число, — это число людей, зараженных одним инфицированным за всё время течения болезни в полностью уязвимой популяции. Очевидно, что это ключевой показатель: если R0 меньше единицы, то эпидемия пойдет на спад, а если больше — то будет расти. Разные заболевания характеризуются очень разными значениями R0 — от 1,3 для гриппа до >10 для кори. Для ближайшего родственника nCoV — SARS — R0 составлял около 3 в начале вспышки и около 0,3 в ее конце [11].
Как измерить R0? Можно строить «традиционные» эпидемические модели и оценивать скорость роста числа заболевших. К сожалению, это трудно сделать точно. Начало вспышки трудно датировать, а в разгар эпидемии многие легкие случаи заведомо не диагностируются; даже скорость диагностики тяжелых случаев может зависеть от пропускной способности системы здравоохранения, например от доступности тест-систем.
Эволюционные методы дают другие способы оценить скорость распространения эпидемии. Основная идея такова: общие статистические характеристики древа, например отношения длин ветвей вблизи корня к длинам ветвей около листьев, должны зависеть от того, сохраняет ли патоген приблизительно постоянную численность или же она растет или убывает. Это связано с тем, что длина ветвей древа, построенного по выборке из некой популяции, определяется численностью этой популяции: чем меньше популяция, тем быстрее «встречаются» друг с другом ветви, поскольку повышается вероятность, что две случайно выбранные особи оказываются близкими родственниками. Растущая вспышка вируса определяется относительно короткими ветвями у корня и относительно длинными — около листьев.
Применение такого анализа к nCoV дает оценки R0 в районе 2 или 3 [7, 8]). Конечно, здесь тоже много предположений, многие из которых заведомо неверны: например, что выборка анализируемых последовательностей более-менее случайна и что на вирус не действует отбор. Но данные, полученные с использованием разных методов, взаимно подтверждают друг друга и позволяют сказать, что R0, по-видимому, находится где-то в диапазоне от 2,2 до 3,3 [12].
Что может быть дальше?
В последние дни появились работы, в которых моделируется развитие эпидемии при различных параметрах [13, 14, 15]. Важно понимать, что эти модели — не прогнозы. Во-первых, точность, с которой оценен R0, совершенно недостаточна. При R0~3 в отсутствие каких-либо мер и без предсуществующего иммунитета вспышка, например, в десятимиллионном городе будет быстрой и острой, будет иметь пик через два-три месяца, и на пике будут одновременно инфицированы десятки процентов населения [15]. Если же R0 ниже двух, то пик растянется на много месяцев и будет смазан (рис. 3).
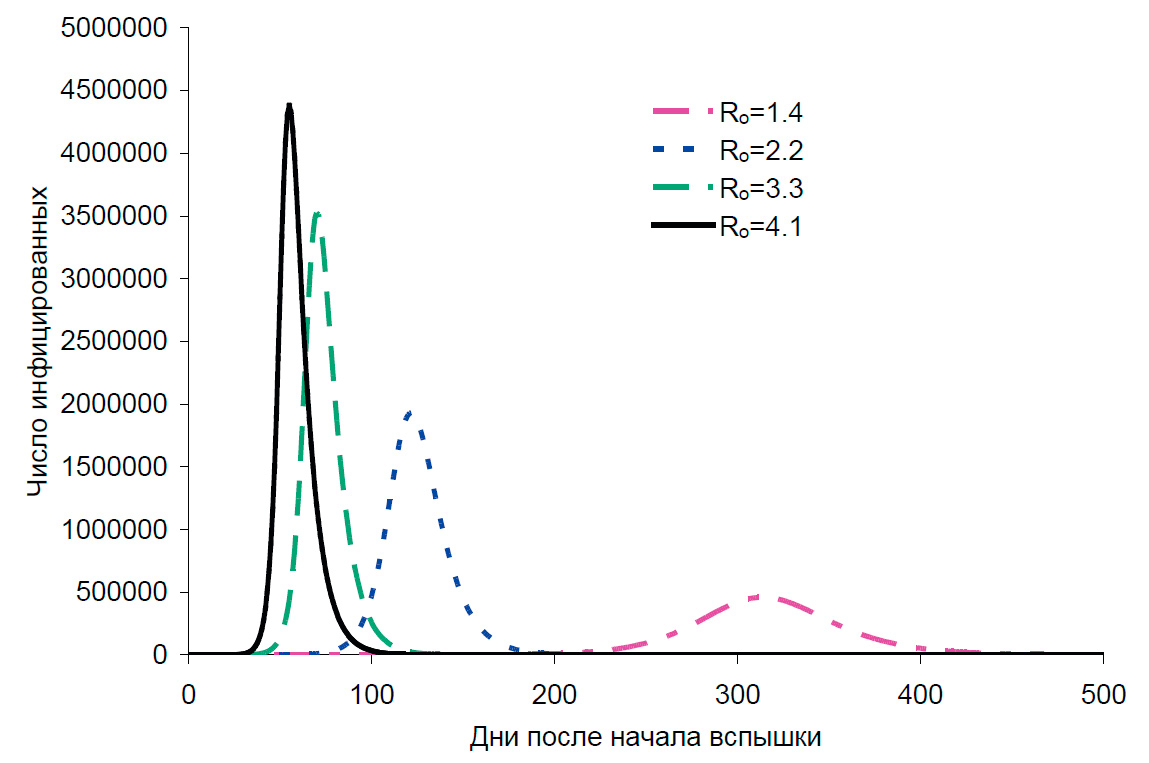
Во-вторых, практически невозможно моделировать такие факторы, как разработка вакцин, эффективность имеющихся лекарственных препаратов (которая остается пока практически неизвестной), а также принимаемые меры по снижению скорости распространения.
В-третьих, сам по себе R0 мало что говорит о том, насколько серьезной окажется эпидемия: R0 для риновирусов, вызывающих обыкновенную простуду, составляет ~6, однако они не являются столь существенной проблемой мирового здравоохранения. Ключевые параметры, остающиеся неизвестными, — это доля тяжелых и смертельных случаев. Составит ли она ~0,1% от общего числа инфицированных, как для ежегодного сезонного гриппа, 2,5%, как для испанки, или 10%, как для SARS? Эти сценарии будут очень разными. Имеющиеся сегодня данные не позволяют сказать, какой из них сбудется.
Георгий Базыкин
(Сколтех, ИППИ РАН)
- Wuhan seafood market pneumonia virus isolate Wuhan-Hu-1, complete genome. (2020).
- Cyranoski D. Did pangolins spread the China coronavirus to people? Nature (2020) doi: 10.1038/d41586-020-00364-2.
- Lu R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 10 (2020).
- Zhou P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 1–4 (2020) doi: 10.1038/s41586-020-2012-7.
- Zhang Z., Shen L. & Gu X. Evolutionary Dynamics of MERS-CoV: Potential Recombination, Positive Selection and Transmission. Sci Rep 6, (2016).
- Coronavirus 2019-nCoV global cases by Johns Hopkins CSSE.
- Rambaut A. Phylodynamic Analysis | 67 genomes | 08 Feb 2020. Virological (2020).
- Bedford T. Nextstrain / narratives / ncov / sit-rep / 2020-01-30 (2020).
- Peck K. M. & Lauring, A. S. Complexities of Viral Mutation Rates. Journal of Virology 92, (2018).
- Poon A. F. Y. et al. Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: an implementation case study. Lancet HIV 3, e231-238 (2016).
- World Health Organization. Consensus document on the epidemiology of severe acute respiratory syndrome (SARS). (2003).
- ncov-R0. Google Docs.
- Read J. M., Bridgen J. R., Cummings D. A., Ho A. & Jewell C. P. Novel coronavirus 2019-nCoV: early estimation of epidemiological parameters and epidemic predictions. medRxiv 2020.01.23.20018549 (2020) doi: 10.1101/2020.01.23.20018549.
- Riou J. & Althaus C. L. Pattern of early human-to-human transmission of Wuhan 2019-nCoV. bioRxiv 2020.01.23.917351 (2020) doi: 10.1101/2020.01.23.917351.
- Wu J. T., Leung K. & Leung, G. M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. The Lancet (2020) doi: 10.1016/S0140-6736(20)30260-9.
- Compartmental models in epidemiology. Wikipedia (2020).
Связанные статьи
- От мозга человека до болезней кораллов (27.03.2018)
- Виктор Малеев: Я дал бы уханьскому коронавирусу еще два месяца (11.02.2020)
- Разоблачен очередной белок, вызывающий рак (09.02.2016)
- Комары против лихорадки (09.05.2017)
- Из Азии в Европу и обратно: степной коридор для чумы (03.07.2018)
- Зомби-гены обонятельных рецепторов (04.07.2017)
- Вирус лишили сладкого (28.08.2018)
- Благо и зло испорченного гена (18.06.2019)
- Эпидемиология и бонистика (28.03.2017)
- Чудо рождения в «Северном Серенгети» (22.05.2018)
Телеграм: t.me/ainewsline
Источник: trv-science.ru