Нейровоспаление: на грани патологического процесса
МЕНЮ
Главная страница
Поиск
Регистрация на сайте
Помощь проекту
Архив новостей
ТЕМЫ
Новости ИИ
Городские сумасшедшие
ИИ в медицине
ИИ проекты
Искусственные нейросети
Искусственный интеллект
Слежка за людьми
Угроза ИИ
ИИ теория
Компьютерные науки
Машинное обуч. (Ошибки)
Машинное обучение
Машинный перевод
Нейронные сети начинающим
Психология ИИ
Реализация ИИ
Реализация нейросетей
Создание беспилотных авто
Трезво про ИИ
Философия ИИ
Генетические алгоритмы
Капсульные нейросети
Основы нейронных сетей
Промпты. Генеративные запросы
Распознавание лиц
Распознавание образов
Распознавание речи
Творчество ИИ
Техническое зрение
Чат-боты
Авторизация
2019-11-26 20:47

Основной упор идет на психиатрические аспекты нейровоспаления, роль нейровоспаления в формировании нервной системы и поддержании гомеостаза ЦНС, а также на спорные моменты влияния на развитие психических и неврологических заболеваний.
Автор не рассматривает роль аутоантител и сценарии клеточной смерти.
Практически любое заболевание ЦНС имеет воспалительный компонент. Трудно обозначить роль воспаления в патогенезе конкретных заболеваний, что особенно важно в контексте отсутствия единого этиологического фактора. Играет ли воспаление основную роль в патогенезе заболевания, просто сопровождает его или же вовсе оказывает нейропротекцию? А может быть, все и сразу? Мы обсудим нейровоспаление, рассматривая его связь с неврологическими и психическими заболеваниями. Разберем основные звенья иммунного компонента ЦНС, защитные, деструктивные и репаративные функции иммунитета.
Рассматривая нейровоспаление с точки зрения психических заболеваний, не лишним будет установить первопричину: это может быть стресс, вызванный психотравмирующей ситуацией, или же локальное воспаление ЦНС, приведшее к столь печальным последствиям. Возможно ли асимптомное (без неврологической симптоматики) нейровоспаление? Будет ли оно охватывать весь мозг или какую-то отдельную зону?
Итак, может ли хронический стресс привести к локальному нейровоспалению? Такой механизм возможен. Эксперимент Caroline Menard et al (2017), проводимый на мышах, демонстрировал локальную проницаемость ГЭБ прилежащего ядра (NAc), обусловленную снижением экспрессии важного белка плотных контактов Cldn-5 (клаудина 5) в ответ на хронический стресс, только у стресс-чувствительных мышей. Впоследствии у этих мышей развивались нейроваскулярные аномалии и повышалась экспрессия CCR-2 (CC-хемокиновый рецептор-2), что сопровождалось локальной инфильтрацией IL-6 паренхимы мозга NAc и симптомами депрессии. Стоит отметить, что длительный прием антидепрессантов (35 дней) способствовал исчезновению симптоматики и нормализации экспрессии белка клаудина-5. В сравнение приводилось посмертное исследование двух когорт пациентов: пациенты первой группы страдали большим депрессивным расстройством (БДР) и совершили суицид, второй — не страдали БДР. Среди лиц с БДР, не принимавших антидепрессанты, уровни экспрессии Cldn-5 были ниже относительно контрольной группы и лиц, принимавших антидепрессанты. Но, к сожалению, можно только предполагать, что прием антидепрессантов мог бы предотвратить некоторые случаи суицида [1].
Развитие воспаления в ЦНС представляет собой мириады реакций, включающие множество путей и разветвлений, а исследования на эту тему зачастую кажутся противоречивыми. Было бы ошибочно просто перечислять пути развития воспаления, механически изображая каждое ответвление в отдельности, искажая полную картину. Поэтому мы будем поэтапно исследовать различные теории и возникающие в связи с ними вопросы, а затем обратимся к механизмам развития нейровоспаления и терапии.
Представления о воспалительной природе психических заболеваний, таких как шизофрения, большое депрессивное расстройство и др. опираются на знания об огромном вкладе кинуренинового пути в развитие как системного, так и локального нейровоспаления, предполагая его роль в патогенезе данных заболеваний. Что такое кинуренин, как он образуется и какова его функция рассмотрим далее.
Кинурениновый путь
Кинуренин-хинолоновый путь — путь превращений аминокислоты триптофана в ходе ее катаболизма, в результате которого образуется несколько крайне важных для понимания нейроиммунологии метаболитов.
Кинуренин (KYN) — метаболит триптофана, образующийся в результате активации 2-х ферментов:
- IDO (индоламин-2,3-диоксигеназа) под влиянием провоспалительных цитокинов, таких как IFN-? (interferon gamma, гамма-интерферон), TNF-? (tumor necrosis factor, фактор некроза опухоли), IL-1,2,6, ROS (reactive oxygen species, активные формы кислорода), LPS (lipopolysaccharide, липополисахарид);
- TDO (триптофан-2,3-диоксигеназа) под влиянием глюкокортикоидов.
Кроме того, с помощью IDO серотонин деградирует до 5-гидроксикинурамина, снижая концентрацию нейромедиатора в синаптической щели [2].
KYN образуется практически повсеместно и имеет способность проникать через гематоэнцефалический барьер (до 60 % кинуренина поступает с периферии, больше всего его образуется в печени). В головном мозге он синтезируется глией и нейронами. Основные «фабрики» по производству метаболитов в головном мозге — микроглия и астроциты. Микроглия синтезирует главным образом хинолиновую кислоту (QUIN), в то время как астроциты (за счет отсутствия фермента KMO) — в основном кинуреновую кислоту (KYNA), также астроциты метаболизируют QUIN [3]..
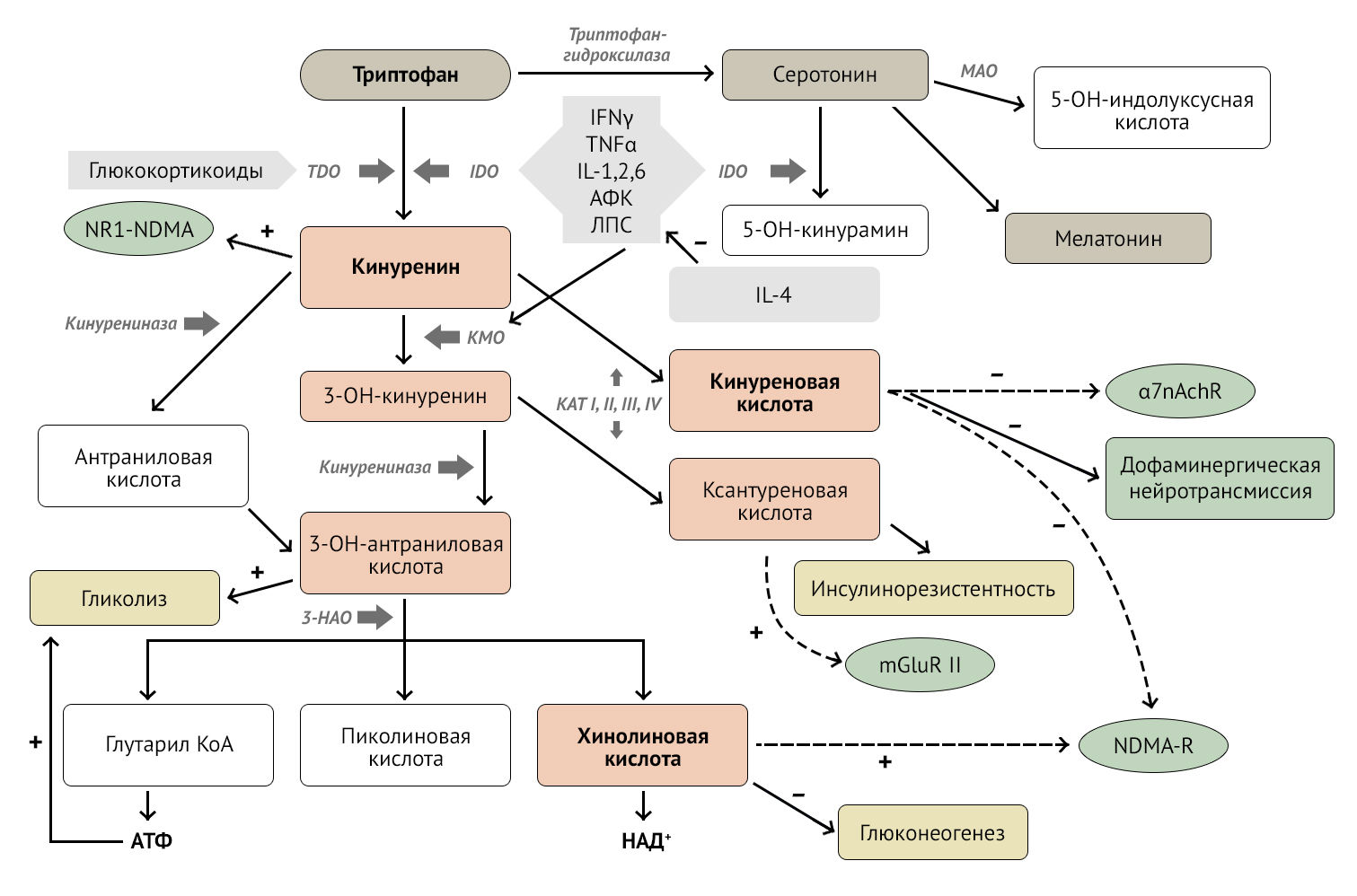
Рисунок 1 | Кинуренин-хинолоновый путь. Описание в тексте [2]
С целью изучения влияния кинуренинового пути на ЦНС было проведено множество исследований, в ходе которых были получены противоречивые результаты — некоторые из них будут рассмотрены ниже.
Кинуреновая кислота
В исследовании Joshua Chiappelli, 2014 сравнивался уровень KYNA в слюне у пациентов с шизофренией и у пациентов из контрольной группы во время стресс-тестов (PASAT — на запоминание и воспроизведение цифр, MTPT — продвижение курсора мыши по тонкой дорожке на мониторе в зеркальном отображении, когда любое отклонение от дороги отбрасывает вас на начало пути), измеряющих тревожность, раздражительность, концентрацию внимания, беспокойство и рабочую память. По итогам, уровень KYNA в слюне значительно увеличился в сравнении с исходным уровнем и уровнем через 20 минут после проведения стресс-теста как у пациентов с шизофренией, так и у контрольной группы (среднее значение — 6,72 нМ [0,65 нМ] против 8,43 нМ [1,05 нМ] соответственно). Пациенты, которые не смогли до конца пройти стресс-тест, определялись как неустойчивые к его прохождению, они показывали значительно более высокий уровень KYNA в слюне, чем стресс-толерантные пациенты или люди из контрольной группы. У стресс-неустойчивых пациентов повышение уровня KYNA соответствовало тяжести клинической симптоматики. Но стоит помнить, что KYNA — неспецифический воспалительный маркер, а значит, заболевания полости рта могли привести к повышенному содержанию этого маркера в слюне, что снижает достоверность результатов исследования [4].
Таким образом, у дистресс-интолерантных участников исследования уровень KYNA был выше относительно стрессоустойчивых лиц. Уровень KYNA коррелировал с тяжестью симптоматики..
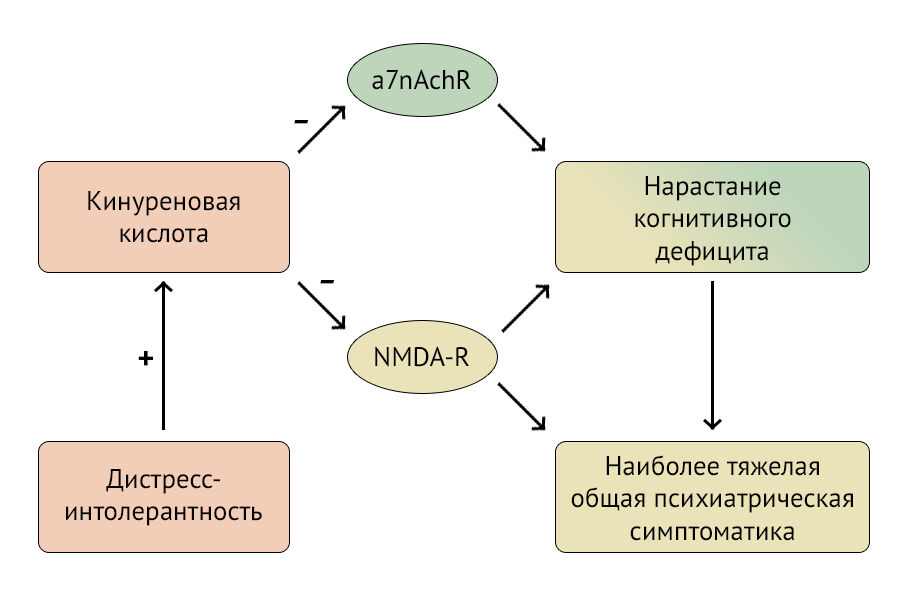
Рисунок 2 | Пути воздействия кинуреновой кислоты на ЦНС
KYNA проявляет нейропротективные свойства, являясь антагонистом глутаматных NMDA-рецепторов, но, блокируя никотиновые рецепторы, она вносит вклад в развитие когнитивного дефицита. Механизм снижения когнитивных функций и ухудшения симптоматики шизофрении представлены в виде схемы на рисунке 2.
В исследовании Chiappelli J (2016) изучалась корреляция отношения концентрации кинуренина к триптофану в сыворотке крови и активности глутаматергической системы белого вещества мозга у пациентов с шизофренией (37 человек) и контрольной группы (38 человек).
У пациентов с шизофренией были снижены уровни триптофана (Trp), а отношение кинуренин/триптофан (KYN/Trp) повышено относительно контрольной группы. Негативная корреляция прослеживалась у пациентов между KYN/Trp и уровнем глутамата в области переднего мозга. Уровни Trp и уровни KYN/Trp никак не коррелировали с тяжестью болезни независимо от приема антипсихотика клозапина или антидепрессантов на момент проведения исследования. Ни Trp, ни KYN/Trp не коррелировали ни с рабочей памятью, ни со скоростью мышления [5].
Таким образом, было выявлено, что деградация триптофана у лиц с шизофренией повышена. Отношение кинуренин/триптофан имеет значимую негативную корреляцию с активностью глутаматергической системы переднего мозга.
Следующее исследование Chiappelli J, 2018, демонстрирует воздействие KYNA на глутаматергическую систему (56 пациентов, больных шизофренией, и 58 человек контрольной группы). В исследование не включались пациенты с неврологическими заболеваниями, такими как эпилепсия, перенесенный цереброваскулярный криз и перенесенная черепно-мозговая травма с проявлениями когнитивных нарушений. Исключались пациенты с активными аутоиммунными или инфекционными процессами, либо ранее подвергшиеся активному лечению по поводу данных заболеваний. Участники с общемедицинской патологией, например, со стабильной гипертонией, из исследования не исключались.
Рассматривалась корреляция уровня KYNA в слюне с уровнем стрессоустойчивости, также при помощи протонной магнитно-резонансной спектроскопии (MRS) измерялась активность глутаматергической системы в передней опоясывающей коре (ACC).
Измерение концентраций KYNA производили перед активацией стрессора, сразу после, через 20 минут после воздействия и через 40 минут от начала эксперимента. Оценку активности глутаматергической системы производили с помощью измерения концентраций глутамата (Glu) и соотношения глутамат/глутамин (Glx). Результаты оказались спорными.
Уровень Glu в ACC был ниже у пациентов с шизофренией, чем у контрольной группы. Уровни Glx существенно не различались. Среди страдающих шизофренией было больше дистресс-интолерантных, чем среди пациентов контрольной группы, но эта разница оказалась незначительной. Уровень KYNA в слюне значимо повысился в ответ на стресс-тест в обеих группах без существенной разницы между исследуемой и контрольной группами.
Уровни Glx, Glu и KYNA статистически значимо коррелировали с проявлениями дистресс-интолерантности у пациентов с шизофренией, в отличие от контрольной группы. После регрессионного анализа уровни Glx показали большую корреляцию с KYNA, нежели Glu.
Ни прием антипсихотиков, ни ИМТ никак не влияли на уровни KYNA и Glx.
Среди больных шизофренией не было значимой корреляции между уровнем KYNA в слюне и нарушением когнитивных функций (по данным тестирования рабочей памяти и скорости мышления), не было выявлено изменений в тяжести симптоматики (по данным результатов тестирования BPRS — краткая оценочная психиатрическая шкала (уровни тревожности, напряженности, враждебности, апатии, нарушенного мышления) или BNSS — краткая оценочная шкала негативной симптоматики (ангедонии, абулии, алогии, дистресса, асоциальности, притупленности аффекта)). Уровни Glx и Glu никак не коррелировали с BPRS или BNSS. Продолжительность болезни прогнозировала снижение уровня Glu после ковариации по возрасту и полу, но не распространялась на уровень KYNA в слюне [6].
Таким образом, можно заключить, что KYNA повышается в ответ на стрессовый фактор. У лиц, больных шизофренией, уровень KYNA негативно коррелирует с активностью глутаматергической системы в ACC. Также уровень Glu коррелирует со скоростью мышления. Повышение KYNA у дистресс-интолерантных пациентов по отношению к стресс-устойчивым не было статистически значимым. Уровень KYNA не коррелировал с тяжестью симптоматики.
Таблица 1 | Изменение метаболизма KYNA при некоторых заболеваниях [7]
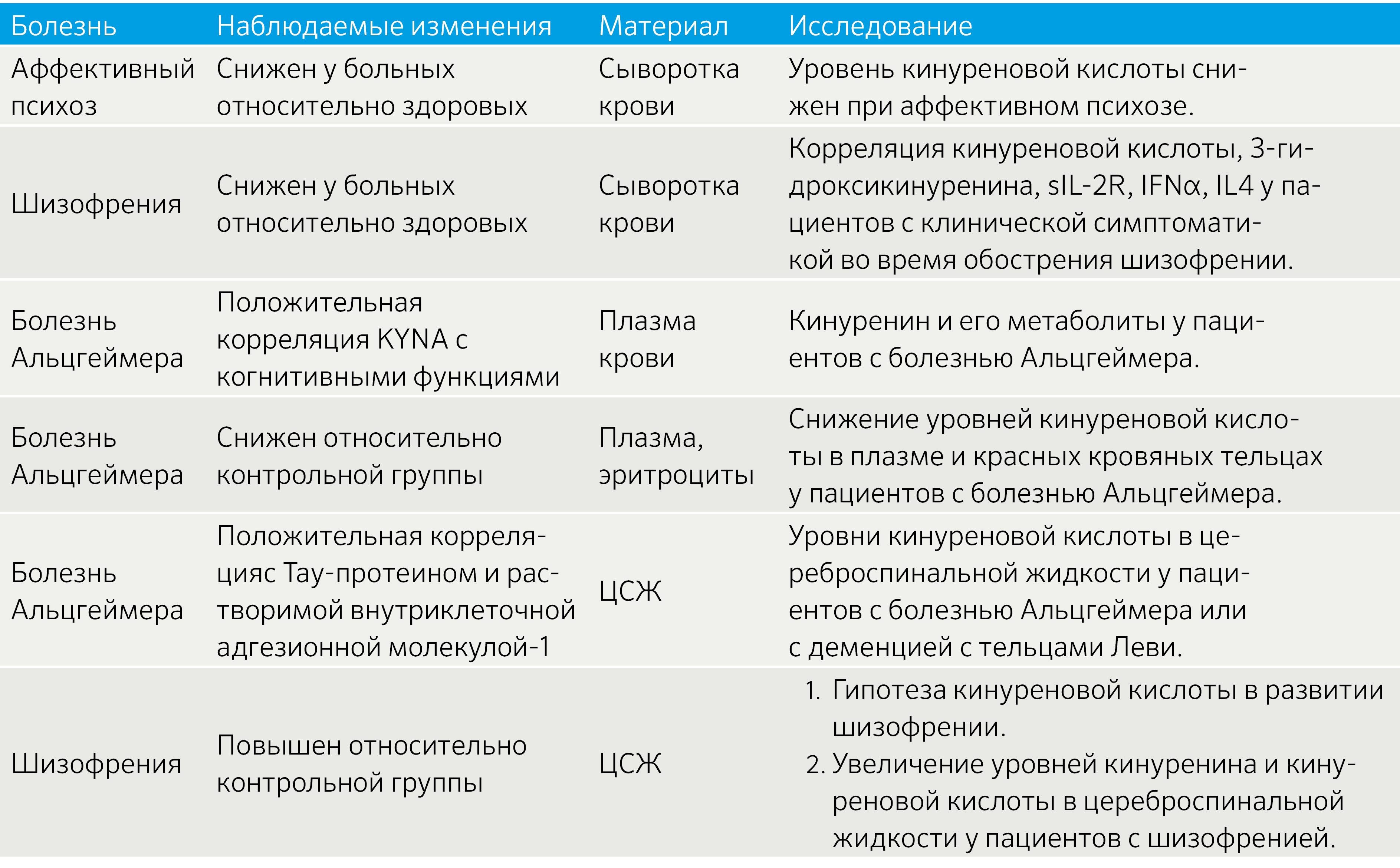
Как следует из приведенных в таблице исследований, качественно не отличающихся от большинства других исследований по данной тематике, результаты спорные. Очевидно, что кинурениновый путь активно вовлечен в формирование и поддержание воспаления, а также воздействует на метаболические процессы в ЦНС. Вместе с этим проявляется причинно-следственная связь между психологическим стрессом и активацией воспаления.
Теперь давайте коснемся места действия нейровоспаления, и в частности кинуренинового пути, и постараемся определить механизмы развития психической и неврологической симптоматики.
Появление симптомов шизофрении при повышении концентрации KYNA связано с тем, что блокирование NMDA-R (NMDA-рецепторов глутамата) ведет к снижению активации глутаматергической системы в мезокортикальном пути, что проводит к нейромедиаторному дисбалансу, реципрокному повышению дофаминового представительства и развитию симптоматики психоза. И в то же время KYNA способствует снижению дофаминовой нейротрансмиссии в стриатуме (полосатом теле). Данное явление обнаружили путем инфузии наномолярных концентраций KYNA непосредственно в стриатум мышей. Происходит это так: KYNA ингибирует альфа-7-никотиновые ацетилхолиновые рецепторы (alpha7nAChRs) на глутаматергических афферентных окончаниях, что ведет к снижению активации AMPA-R (AMPA-рецепторов глутамата) на дофаминергических нейронах [8]. Но ведь именно дисбаланс в сторону повышения дофаминергической трансмиссии в дорсальной области стриатума лежит в основе современной дофаминовой теории возникновении шизофрении [9]. Это приводит нас к выводу о том, что повышенная концентрация KYNA усугубляет негативную симптоматику (за счет блокады никотиновых рецепторов) и блокирует NMDA-рецепторы в мезокортикальном пути, но также одновременно снижает дофаминергическую нейротрансмиссию в стриатуме, что, казалось бы, должно способствовать уменьшению продуктивной симптоматики. Все это требует дополнительного изучения влияния KYNA на проявления шизофрении. Тем не менее подход к лечению индуцированной кетамином симптоматики психоза модуляторами кинуренинового пути уже представлен, однако пока только на мышах.
Данное исследование базируется на другом эксперименте, в котором демонстрируется повышенная концентрация маркеров оксидативного стресса, таких как малондиальдегид, у пациентов с шизофренией.
Исследование, которое мы сейчас рассмотрим, проводилось на мышиной модели шизофрении (индуцированный кетамином психоз) с применением ингибиторов IDO-2,3 и TDO-2,3. Введение кетамина мышам сопровождалось появлением спонтанной локомоторной активности (аналог продуктивной симптоматики психоза у мышей). Однако ингибиторы IDO-2,3, TDO-2,3 и KMO были способны устранять данные проявления. В дополнение, IDO-ингибитор предотвращал перекисное окисление липидов и снижал уровни карбонильных белков (маркеры оксидативного стресса) в префронтальной коре (PFC) и гиппокампе. TDO-ингибитор делал то же самое в стриатуме и гиппокампе. Кроме того, он повышал уровень SOD (супероксиддисмутаза — антиоксидант, нейтрализующий супероксидные радикалы) в стриатуме и активность CAT (холин-ацетилтрансфераза — фермент, участвующий в образовании ацетилхолина) в гиппокампе. KMO-ингибитор не смягчал повреждение от перекисного окисления липидов, но повышал активность SOD в гиппокампе мышей и активность CAT в стриатуме, а также снижал концентрации карбонильных белков у мышей, которым производили инъекции кетамина [10].
Изучая кинурениновый путь, особое внимание стоит уделить метаболиту под названием хинолиновая кислота (QUIN). Она является агонистом NMDA-R, а увеличение ее концентрации может привести к эксайтотоксичности и нейродегенерации. Прочесть статью, рассматривающую различные теории шизофрении, в том числе нейродегенеративную, вы можете здесь.
QUIN нарушает активацию FGF2-ERK-пути и приводит к дестабилизации цитоскелета астроцитов путем изменения редокс-потенциала и повреждения щелевых контактов в случае нахождения в смешанной культуре клеток (астроцит + нейрон + микроглия). Fibroblast Growth Factor-2 — фибробластоподобный фактор роста 2 — синтезируется в основном в астроцитах в ответ на стрессовые сигналы, он чрезвычайно важен в ходе нейрогенеза, способствует репарации ЦНС при повреждении, а также модулирует воспалительные пути, которые регулируют микроглиальную активацию. FGF играет важную роль в нейровоспалении, а FGF2-ERK1/2 путь рассматривают в качестве нейропротективного (против глутамат-опосредованной эксайтотоксичности).
Через активацию ERK-1/2 (extracellular signal-regulated kinase-1 и 2 — киназа, регулируемая внеклеточными сигналами 1 и 2) и PKC (протеинкиназа C) FGF влияет на фосфорилирование структурных белков — коннексинов (Cx), ответственных за формирование, расформирование и функциональную активность щелевых контактов (Gap junctions, Gjs)..
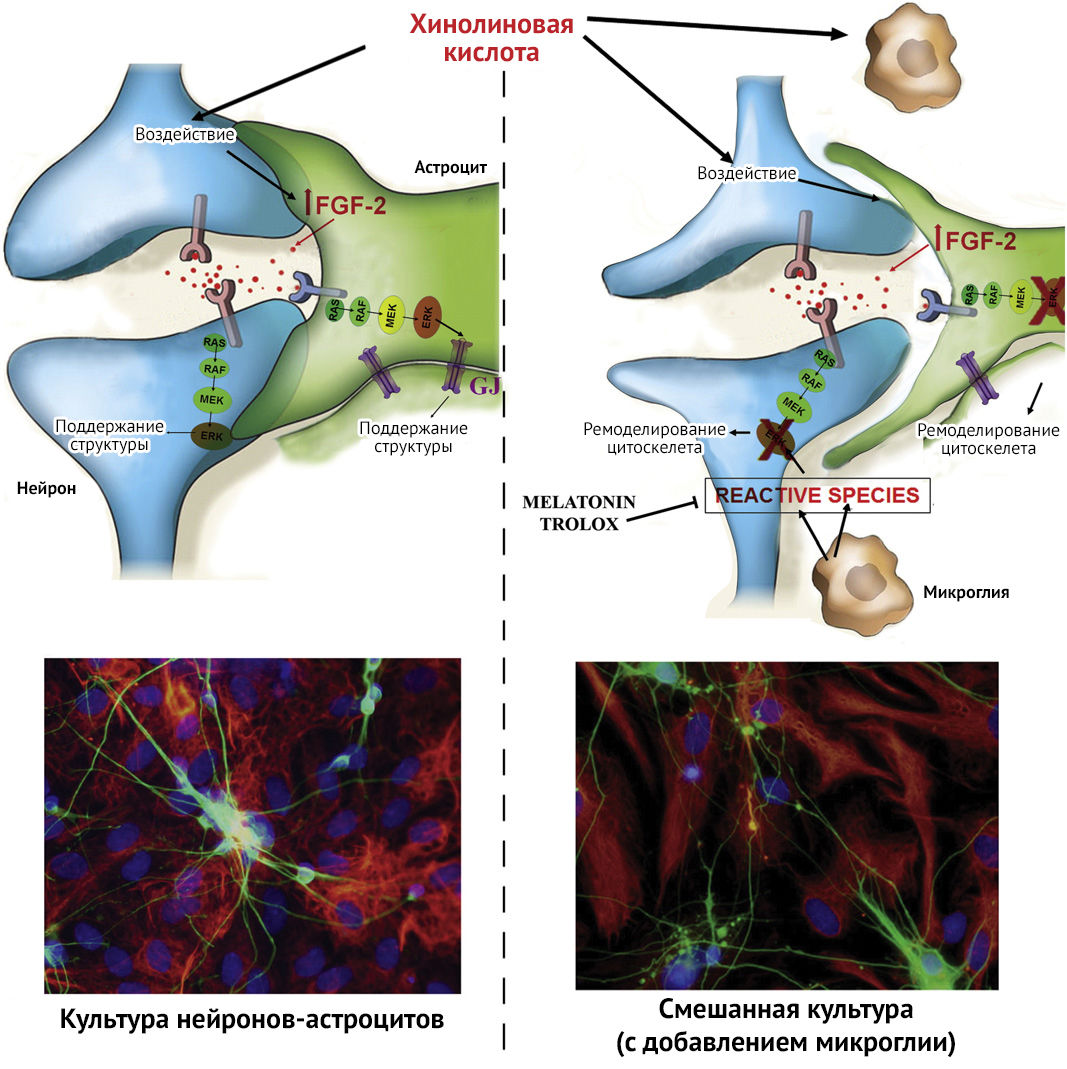
Рисунок 3 | Эффект воздействия хинолиновой кислоты (QUIN) на культуру нейронов-астроцитов (coculture — 1) и на смешанную культуру (с добавлением микроглии) (mixed culture — 2) [12]
Gjs — межклеточные каналы, обусловливающие взаимодействие между клетками, способствуют передаче малых молекул и ионов (например, Ca2+, глутамата, метаболитов и т. д.) между цитоплазмой соседних клеток. Они состоят из двух межклеточных полуканалов, которые формируют один щелевой контакт. Полуканалы у позвоночных могут быть представлены двумя типами белков: Cx и панексинами. Функциональная интеграция различных типов клеток чрезвычайно важна для поддержания сигнального гомеостаза в ЦНС. Так, дисфункция этой коммуникативной системы клеток способна негативно отразиться на жизнеспособности и функционировании нейронов.
Астроциты активно взаимодействуют через Gjs, распространяя скоординированные сигналы различным клеткам и формируя электрически и метаболически связанные единицы. Активность Cx-43 — основного Cx, экспрессирующегося в астроцитах, может подавляться воспалительными цитокинами, приводя к нейродегенерации и нейровоспалению. С другой стороны, нейроны могут регулировать экспрессию Cxs через высвобождение биологически активных молекул, которые способны активировать астроцитарные рецепторы и модифицировать активность и/или экспрессию Gj каналов.
Основной поставщик цитокинов в ЦНС — микроглия. Микроглиальные клетки могут активироваться двумя путями: классическим — M1 (когда микроглия экспрессирует провоспалительные медиаторы) и альтернативным — M2 (противовоспалительные, нейропротективные медиаторы). Кроме того, M1-активация может рассматриваться как основной источник генерации ROS в ЦНС, а ROS приводят к оксидативному стрессу и вовлечены в патогенез тяжелых нейродегенеративных расстройств. Активация по M1 либо по M2 пути будет зависеть от множества сигналов, исходящих из поврежденного окружения мозга, особенно из квадрисинапса.
Квадрисинапс состоит из пре- и постсинаптического нейронов, астроцита и микроглиальной клетки и представляет собой разнообразие новых коммуникаций, которые при M1-активации приводят либо к дисфункции, либо к разрыву межнейронных взаимосвязей и развитию хронического нейровоспаления.
Напомним, что одна из причин, по которой кинурениновый путь может привести к нейродегенерации и нейровоспалению — хинолиновая кислота (QUIN). QUIN — агонист NMDA-рецепторов, гиперактивация которых приводит к нейрональной и астроцитарной эксайтотоксичности, гиперпродукции свободных радикалов, агрессии микроглии. Также QUIN нарушает глюконеогенез, что крайне губительно для мозга в случае гипоксии. Для большей убедительности отметим, что хинолиновую кислоту в исследовательских целях вводят в стриатум мышам для создания мышиной модели болезни Хантингтона, а это приводит к гиперпродукции воспалительных цитокинов TNF-? и IL-6 [11].
В исследовании влияния QUIN на цитоскелет было выявлено, что в трисинаптическом варианте (пре- и постсинаптические окончания нейронов + астроцит) введение хинолиновой кислоты не вызывает негативных эффектов ввиду наличия астроцит-нейрональной нейропротективной связи (рис. 3 (1)), в отличие от отдельной культуры клеток нейронов или астроцитов, структура которых повреждалась в ответ на введение QUIN. Это объясняется слаженной работой щелевых контактов, чья деятельность напрямую зависит от структурных белков Cx-43, фосфорилирование которых происходит при активации пути FGF2-Erk. Однако в квадрисинапсе происходит активация микроглии, формирование амебоподобных клеток, снижение объема цитоплазмы астроцитов и ветвление нейронов. Хотя уровни FGF2 и были повышены, это не способствовало активации MEK-Erk пути (MEK относится к митоген-активируемым протеинкиназам (MAPK)), концентрация Cx-43 была снижена, что демонстрировало основной негативный эффект от QUIN, а именно — блокирование Gjs (рис. 3 (2)).
Дезорганизация цитоскелета, вызванная введением QUIN в квадрисинаптическую культуру, предотвращалась добавлением антиоксидантов мелатонина и Тролокса (водорастворимый аналог витамина Е). Это наводит на мысль о причинной связи «QUIN — оксидативный стресс — нарушение активации тирозинкиназного рецептора — реорганизация цитоскелета» [12].
Чтобы примерно разобраться, чем конкретно отличаются пути активации M1 от M2, посмотрим на картинку ниже..
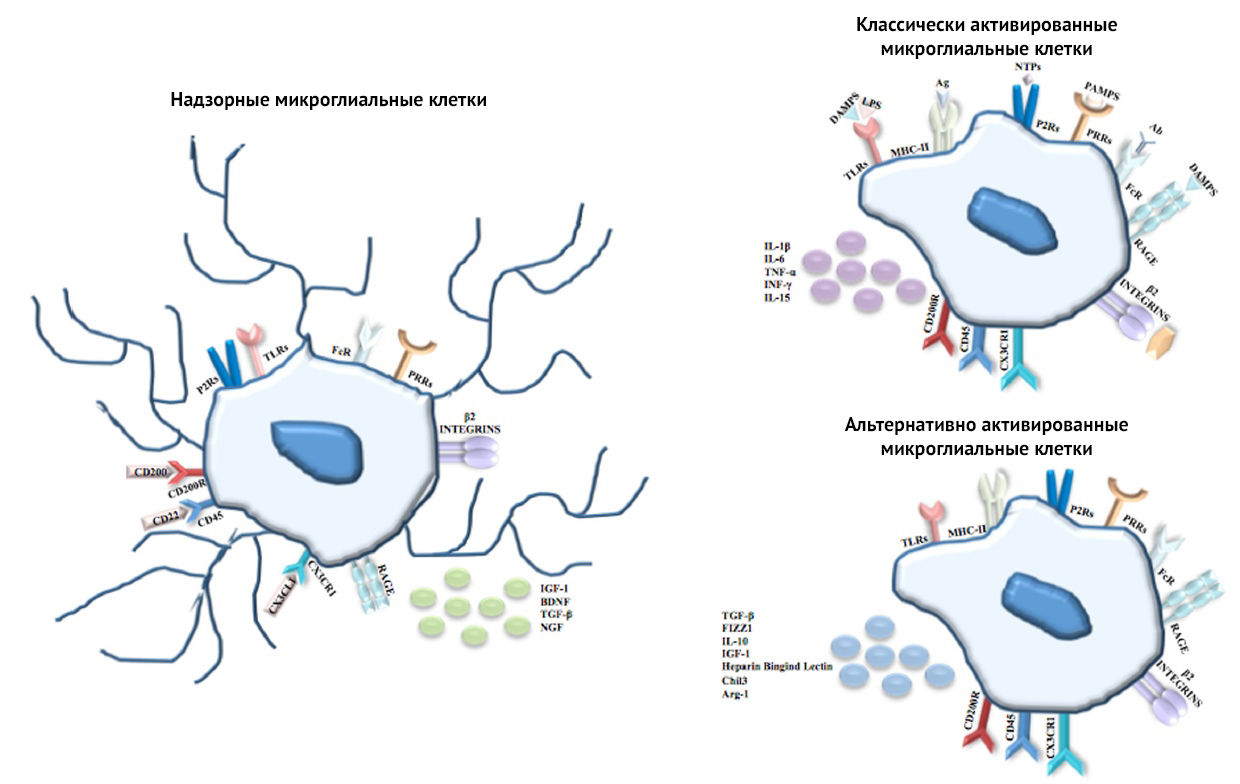
Рисунок 4 | Микроглиальные клетки
В физиологическом состоянии и в ходе развития ЦНС клетки микроглии как клетки иммунного надзора имеют ветвистую морфологию и, соблюдая моноцитарно-макрофагальную династию, экспрессируют на своей мембране поверхностные ?2-интегрины (CD11?, CD11?), Fc-рецепторы, и PRRs (паттерн-распознающие рецепторы), включая TLR (Toll-подобные рецепторы), NLR (Nod-подобные рецепторы), RLR (RIG-подобные рецепторы), CTLR (лектиновый рецептор типа C), P2Rs (P2-пуринорецепторы) и RAGE (рецепторы продуктов длительного гликозилирования). Состояние надзорной клетки (состояние покоя, М0) обеспечивается через ингибиторные сигналы, возникающие между CD200 и CD200R, CD22 и CD45, CX3CL1 (Фракталкин) и CX3CR1, и характеризуется секрецией IGF-1 (инсулиноподобный фактор роста-1), BDNF (нейротрофический фактор мозга), TGF-? (трансформирующий ростовой фактор бета) и NGF (фактор роста нервов). При распознавании PAMPs (патоген-ассоциированные молекулярные паттерны), таких как ЛПС, вирусные частицы, компоненты бактериальной клетки, ядерные белки DAMPs (ассоциированные с опасностью/повреждением молекулярные паттерны или просто алармины) — HMGB1 (B1-белок с высокой электрофоретической подвижностью, амфотерин) и S100B (белок из группы кальций связывающих белков), NTPs (нуклеозидтрифосфаты) или белки комплемента, микроглия переходит в состояние классической активации (M1-фенотип), которое характеризуется сверхрегуляцией по MHC II и секрецией провоспалительных цитокинов, таких как IL-1?, IL-6, TNF-?, IFN-? и IL-15. Однако чтобы уменьшить повреждение нейронов, микроглия после активации иммунного ответа переходит в состояние активации по альтернативному пути (M2-фенотипа), характеризующееся релизом аргиназы-1 (способствует репарации ткани), гепарин-связывающего лектина и хитиназы-3 (предотвращает деградацию внеклеточного матрикса в зоне воспаления и способствует формированию самого матрикса), а также секрецией противовоспалительных цитокинов — IL-10, TGF-? и IGF-1 [13].
Для наглядной демонстрации нейрон-микроглиального взаимодействия приведены рисунки 5 и 6. Поддержка надзорной функции микроглии, ее противовоспалительного фенотипа обусловлена непосредственным межклеточным контактом между CD200 и CD200R (но не обязательно в отношении CX3CL1 и CX3CR1, когда CX3CL1 может принимать форму растворимого лиганда и взаимодействовать со своим рецептором без непосредственного контакта между нейроном и микроглиальной клеткой).
.
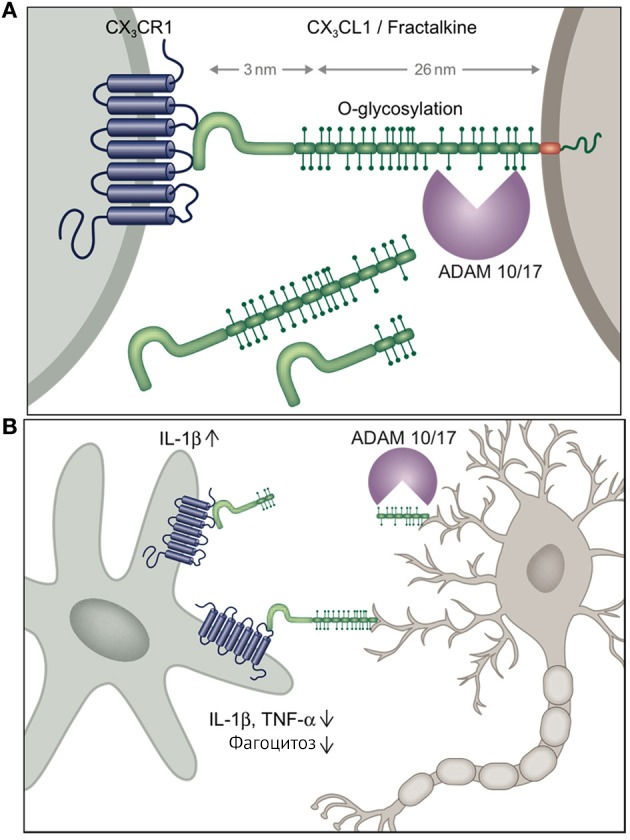
Рисунок 5
CX3CL1 подвергается протеолитическому разрезанию металлопротеиназой ADAM10, обеспечивая взаимодействие лиганда с рецептором на расстоянии. Вне ЦНС CX3CL1 имеется в клетках кишечного эпителия и эндотелиальных клетках, а CX3CR1-рецептор экспрессируется на натуральных киллерах, Т-клетках и миелоидных мононуклеарах. Однако в ЦНС CXC3CL1 экспрессируется только на нейронах, а CX3CR1 — на микроглии.
Взаимодействие лиганда с рецептором приводит к снижению провоспалительной функции через подавление секреции IL-1?, TNF-?, а также супрессию фагоцитарной функции [14].
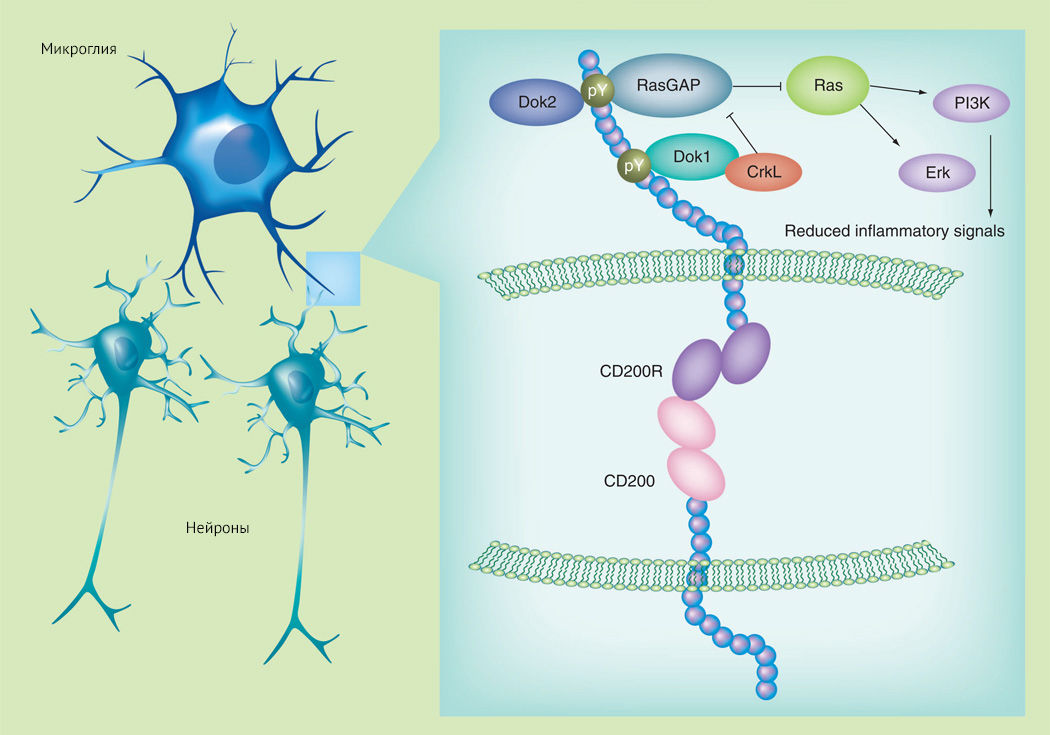
Рисунок 6
В ЦНС CD200 экспрессируется в основном на нейронах, а CD200R — на микроглиальных клетках. Основной механизм действия CD200R проявляется активацией Dok2 (downstream of kinase-2, p62) и RasGAP (белок, активирующий ГТФазу Ras), что приводит к подавлению активации Ras.
С возрастом количество CD200 снижается, что коррелирует с увеличением концентрации провоспалительных цитокинов и экспрессией провоспалительных маркеров на микроглиальных клетках [15].
Добавление в культуру микроглиальных клеток с ?-амилоидом нейронов снижает концентрацию провоспалительных цитокинов, вероятно, через CD200 [14]. Однако говорить об однозначно положительном результате такого взаимодействия не приходится. К примеру, противовоспалительный CD22, снижающий концентрацию провоспалительных цитокинов и подавляющий фагоцитирующую функцию микроглии, также подавляет фагоцитоз миелинового мусора, ?-амилоида, ?-синуклеина. Блокада CD22 приводит к нормализации микроглиального гомеостаза. Концентрация нейропротективного CD22 увеличивается с возрастом, что выводит на первый план негативные последствия антифагоцитарной активности в виде белковых агрегатов, миелинового мусора, развития нейродегенерации [16].
Кроме того, в посмертных образцах головного мозга людей, страдавших болезнью Альцгеймера, в микроглии выявлялась повышенная экспрессия генов, кодирующих C1q, C3 и C4 белки системы комплемента, что, несомненно, говорит о большом вкладе воспаления в развитие данного заболевания [17]. Однако неясно, сказывается ли экспрессия данных генов негативно на развитии заболевания. C1q является фактором активации системы комплемента по классическому пути, его физиологическая функция включает в себя элиминацию антител и апоптотических тел. Дефицит факторов C1q, C3 и C4 может быть одной из причин развития аутоиммунных заболеваний [18, 19]. Также вероятно, что повышение концентрации компонентов системы комплемента при болезни Альцгеймера связано с недостаточным фагоцитозом ?-амилоидных фибрилл.
Негативное влияние системы комплемента проявляется только при ее гиперфункции, когда запускается положительная обратная связь, при которой воспаление потенцирует воспаление. Белки комплемента могут попасть в ЦНС при нарушении проницаемости ГЭБ либо могут быть синтезированы микроглией и нейронами (в меньшей степени). Система комплемента активируется в ответ на появление амилоидных фибрилл, что через опсонизацию в дальнейшем приводит к усилению фагоцитарной активности микроглии (с целью утилизации ?-амилоида). Однако это же приведет к смещению фенотипа микроглии в сторону М2-активации и секреции провоспалительных цитокинов, кроме того, система комплемента опосредует хемотаксис и привлечение иммунокомпетентных клеток, активацию B-клеток, формирование мембраноатакующего комплекса и аутоагрессию [20].
Вернемся к описанию М0, М1 и М2 фенотипов микроглии (рис. 4). Основная роль в дифференцировке «нативного» макрофага принадлежит паттерн-распознающим рецепторам и собственно паттернам, активирующим их (DAMP или PAMP), а также IFNy (гамма-интерферон). Кроме того, значимую роль в развитии макрофага играют белки семейства HIF (индуцированный гипоксией фактор). Но обо всем по порядку.
Таблица 2 | Паттерн-распознающие рецепторы и их лиганды [21]
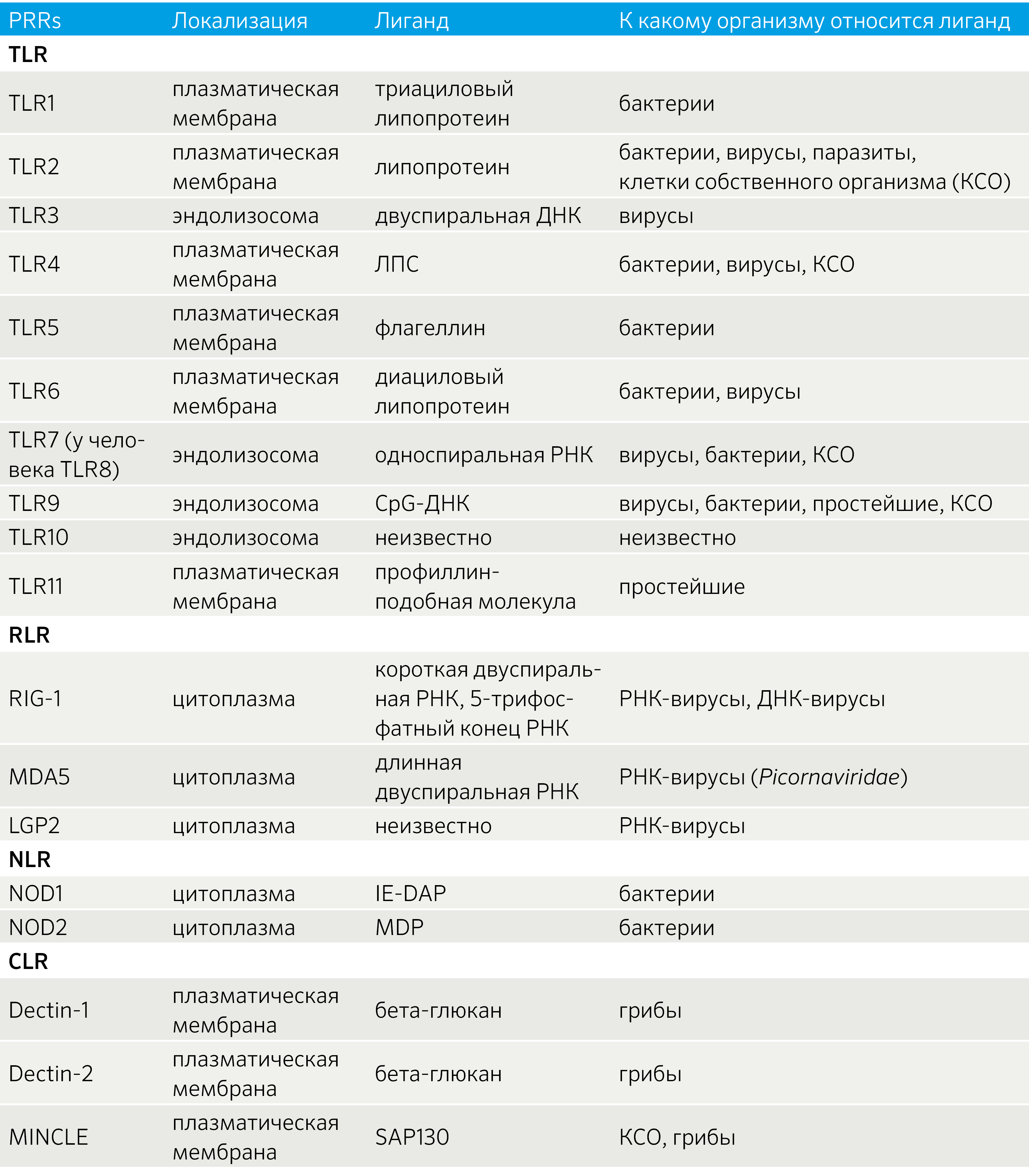
Паттерн-распознающие рецепторы являются сенсорной системой, позволяющей активировать иммунный ответ на потенциально опасные компоненты внутренней среды самого макроорганизма, либо в случаях высвобождения данных веществ, связанных с асептической травмой или гипоксией (DAMP), либо на определенные компоненты бактериальных, вирусных, грибковых, паразитарных организмов (PAMP) (табл. 2). Активация PRR необходима для осуществления иммунного ответа, хроническая же активация может приводить к развитию аутоиммунных и аутовоспалительных заболеваний..
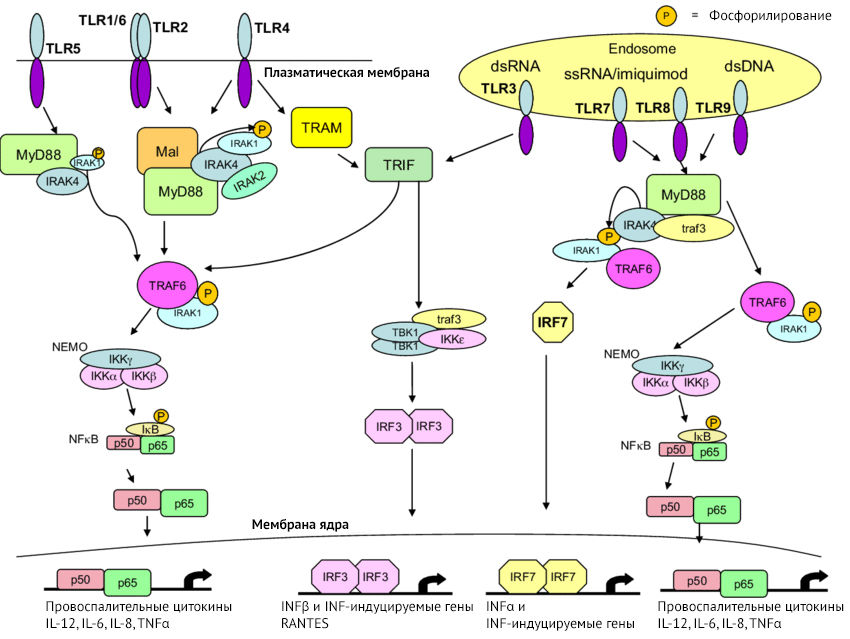
Рисунок 7 | Активация Toll-подобного рецептора
Дабы не перегружать вас информацией, отметим, что PRR работают в содружестве друг с другом. К примеру, для секреции IL-1? необходима активация NF-kB (ядерный фактор каппа-B — важный для синтеза провоспалительных цитокинов транскрипционный фактор) через TLR, после чего происходит формирование про-IL-1?. Далее для разрезания про-IL-1? и превращения его в IL-1? необходимо синтезировать каспазу-1, что и происходит после активации NLR и формирования инфламмасом.
Большинство клеток в ЦНС экспрессирует PRR. Для NLR отмечается возможность формирования инфламмасом в астроцитах в ответ на выброс АТФ или насыщенных свободных жирных кислот, в микроглии — из-за ?-амилоида, с возможным развитием пироптоза (вид клеточной смерти).
При спинномозговой травме формируется глиальный рубец в результате астроглиоза. Рубец является механической и молекулярной преградой для успешной регенерации аксонов. Связывают это с активацией Rig1 и MDA5 (melanoma differentiation associated protein 5) — рецепторов группы RLR, что приводит к возрастанию концентраций глиального фибриллярного кислого белка (GFAP) и виментина в астроцитах. В эксперименте на фоне контузионной травмы шейного отдела у крыс экспрессия Rig1 и MDA5 повышалась в течение 6 часов после повреждения, приводя к увеличению секреции цитокинов IFN I типа: IFNa и IFN?. Установлено, что RLR-сигналинг в астроцитах может быть ингибирован митохондриальным убиквитин-лигазным белком E3, что приводит к идее использования механизма активации RLR для предотвращения формирования глиального рубца [21].
В формировании глиального рубца участвует процесс воспаления. Нарушение миелинизации и ремиелинизации также обусловлено воспалительным процессом. Предполагалось, что активация TLR будет вносить вклад в прогрессирование демиелинизации. Однако данные оказались совершенно противоположными.
TLR-4-сигналинг индуцировал формирование олигодендроцитов в нетронутом спинном мозгу, а отсутствие TLR-4 у мышей приводило к обеднению белого вещества и худшему восстановлению после травмы спинного мозга. У TLR4-дефицитных мышей потеря олигодендроцитов сопровождалась сниженной экспрессией ферритина, которая регулировалась через TLR4 и была необходима для эффективного связывания железа и предотвращения ферроптоза. У этих мышей была нарушена миграция клеток-предшественников олигодендроцитов, а также снижена экспрессия факторов, необходимых для пролиферации и роста, включая IGF-1, FGF-2 (фактор роста фибробластов), IL-1? и PDGF-A (фактор роста тромбоцитов А) [22].
Как уже упоминалось, RAGE (рецепторы продуктов конечного гликозилирования) входят в число PRR. Однако сам RAGE является одним из рецепторов большого семейства: AGE-рецепторного комплекса (AGE-R1/OST-48, AGE-R2/80K-H, AGE-R3/galectin-3) и скавенджер-рецепторов (SR-A; SR-B: CD36, SR-BI SR-E: LOX-1; FEEL-1; FEEL-2). Экспрессия этих рецепторов зависит от клеток и тканей, а разнообразие их функций очень велико: от активации иммунного ответа до регуляции метаболизма клетки, роста и развития, межклеточных взаимодействий. Лигандами RAGE являются AGE (конечные продукты гликозилирования) — белки, длительное время подвергавшиеся гликозилированию, что изменило их структуру и нарушило процесс их фагоцитоза и утилизации, а также HMGB, S100B и ?-амилоидные фибриллы. Результат активации RAGE зависит от лиганда и клеток, на которых RAGE экспрессируется: активация на клетках микроглии будет приводить к развитию иммунного ответа по М1-фенотипу, однако при небольших концентрациях HMGB1 и S100B, присоединившись к рецепторам RAGE на нейронах, приведут к аксональному росту [23].
Дифференциация в сторону воспалительного фенотипа микроглии идет и при участии IFN?. Происходит это в случае инфицирования клеток. При активации гамма-интерфероновых рецепторов 1 и 2 (IFN?R) запускаются NF-kB (ядерный фактор каппа B) и STAT 1 (преобразователь сигналов и активатор транскрипции 1), что в свою очередь повышает экспрессию CD86 и MHC II, провоспалительных цитокинов, хемокинов и т. д. IFN? далеко не всегда действует самостоятельно, зачастую активация через PRR и IFN?R идет параллельно (рис. 8) [24]..
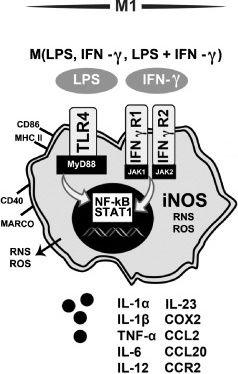
Рисунок 8 | Характеристика М1-активированной микроглиальной клетки
Что касается HIF-активации, дело обстоит следующим образом.
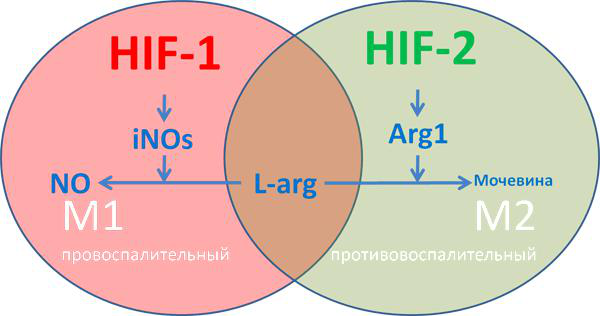
Рисунок 9 | Влияние HIF на дифференциацию макрофага
М1-макрофаги характеризуются «провоспалительным» фенотипом, преимущественно гликолитическим типом метаболизма, синтезом провоспалительных цитокинов, формированием реактивных форм азота и кислорода. М2-макрофаги характеризуются «противовоспалительным» фенотипом, преимущественно окислительным фосфорилированием, синтезом аргиназы ARG1 и противовоспалительных хемокинов типа IL-10, IL-13, IL-4, факторов роста TGF? и IGF. У iNOs и ARG1 имеется единый субстрат — L-аргинин, содержание которого в межклеточной среде ограничено (рис. 9) [25]. Аргиназа конвентирует L-аргинин в орнитин, обладающий репаративным действием, индуцибельная NO-синтаза преобразует аргинин в реактивные формы азота.
Стоит понимать, что М1 и М2 фенотипы — упрощенное обозначение провоспалительной и противовоспалительной активации микроглии. На самом деле функции микроглии очень разнообразны и зависят от морфологии и расположения: клетки микроглии белого вещества — с вытянутыми тельцами и отростками, тянущимися в сторону волокон белого вещества; клетки микроглии желудочков мозга меньше микроглиальных клеток серого и белого вещества, с короткими, редкими аксонами; клетки микроглии серого вещества — самые ветвистые.
Кроме того, микроглиальные клетки разделяют по механизмам их активации и паттернам экспрессии белков, молекул, цитокинов (рис. 10). Так, М2-клетки подразделяются еще на 3 фенотипа: М2a, М2b, M2c..
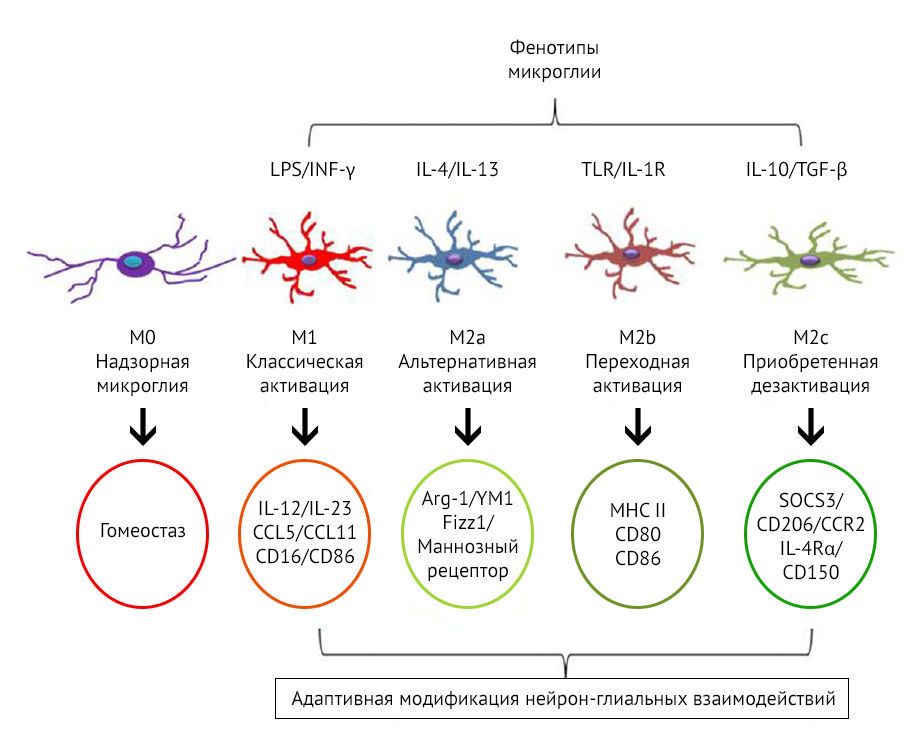
Рисунок 10 | Фенотипы М1 и М2 [26]
К основным функциям M2a относятся репарация и регенерация, M2b — иммунная регуляция, M2c — нейропротекция и секреция некоторых противовоспалительных цитокинов.
На вопрос, в регенерацию чего вовлечена M2a микроглия, можно ответить так: большое депрессивное расстройство имеет множество нейробиологических субстратов, одним из которых может являться снижение регенерации нейронов в зубчатой извилине гиппокампа. Поляризация микроглии с М1 на М2, как показывают исследования на лабораторных животных, нормализует нейрорегенерацию и снижает симптоматику депрессии [26].
Что касается методов, направленных на поляризацию микроглии, in vitro это делается при помощи добавления к культурам определенных цитокинов: к M1 — IFN?, LPS; к M2 — TGF?, IL-10, IL-4, активация PPAR? (активатор пролиферации пероксисом, основная роль которого состоит в ингибировании провоспалительного сигналинга STAT — NF-kB (рис. 11)) [27].
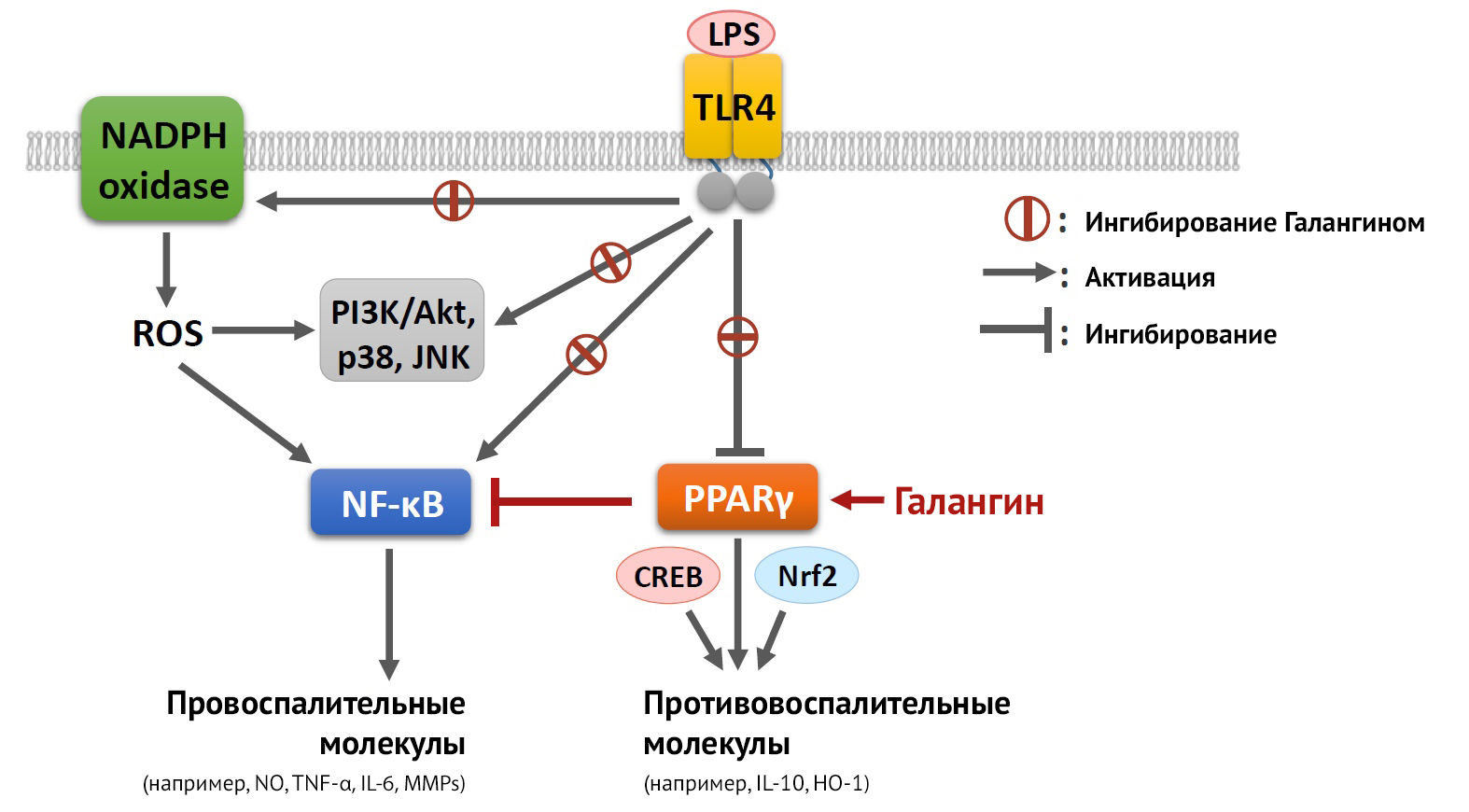
Рисунок 11 | Реализация противовоспалительного механизма действия PPARy после активации галангином
Кроме того, поляризацию макрофагов и микроглии удается осуществить при помощи молекул железа. М1-микроглия характеризуется повышенным поглощением железа, блокированием высвобождения связанного с белком трансферрина, образованием реактивных форм кислорода и азота. М2, напротив, характеризуется снижением поглощения железа, обусловливая захват большего количества железа другими клетками, что необходимо для их репарации (рис. 12) [28].
В настоящее время данные вопросы часто поднимаются при обсуждении онкологических заболеваний и необходимости провоспалительной поляризации макрофага с целью формирования противоопухолевого иммунитета. Для этого разрабатываются препараты, приводящие к накоплению железа в тканях опухолевого окружения, например, магнетит.
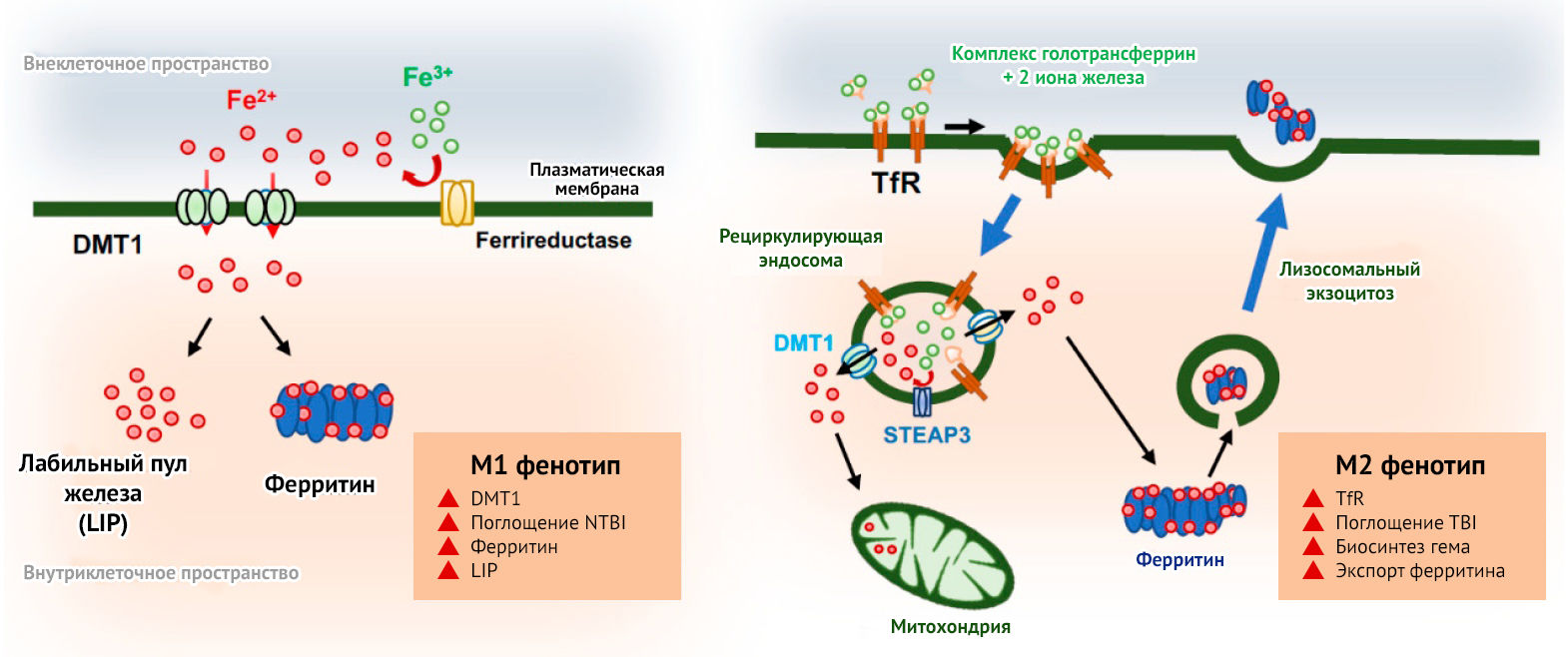
Рисунок 12 | Метаболизм железа и поляризация микроглии
DMT1 — транспортер двухвалентного металла-1; NTBI — не-трансферрин связанное железо; TBI — трансферрин связанное железо; TfR — рецептор трансферрина [28].
Оксидативный стресс, поляризация М1-макрофагов, секреция провоспалительных цитокинов, образование реактивных форм кислорода и азота через реакцию Фентона, ферроптоз (вид клеточной смерти, одним из обязательных рычагов активации которого является избыточное накопление внутриклеточного железа) и ингибиторы антиоксидантов — все это имеет большое значение в развитии нейродегенеративных и аутоиммунных заболеваний, а также повреждений в результате геморрагического инсульта и реперфузионных постишемических поражений мозга [29].
Астроциты также вносят немалый вклад в формирование фенотипа микроглии и нейровоспаление.
Сама по себе астроглия может влиять на воспаление, потенцируя либо ослабляя его секрецией цитокинов, воздействующих на микроглию и проницаемость ГЭБ (рис. 13) [30]..
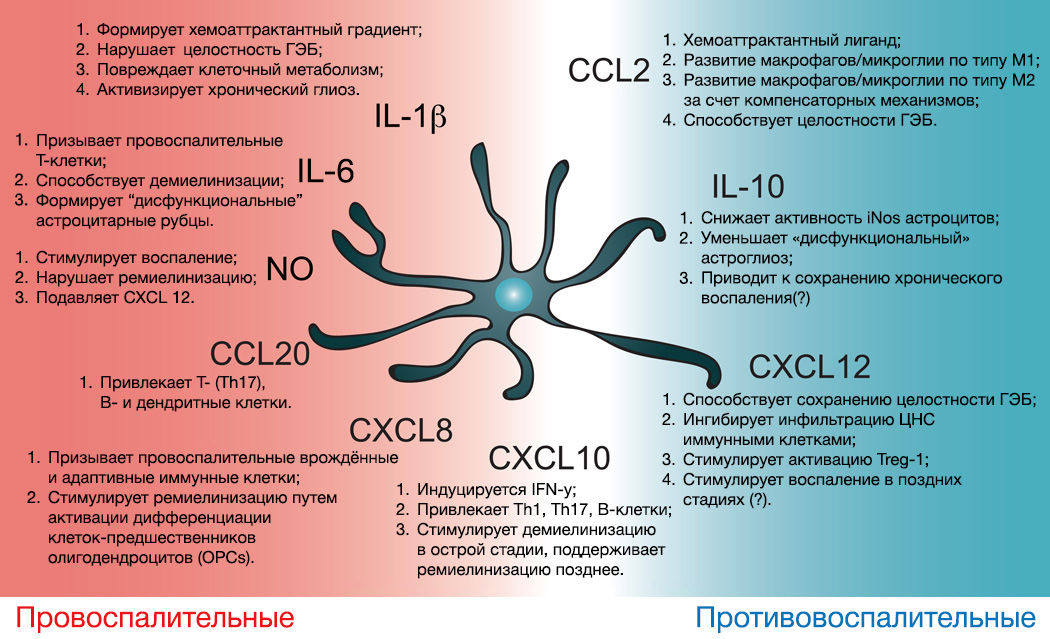
Рисунок 13 | Иммунная активность астроцитов
Помимо представителей глии, в иммунной регуляции участвует и сам нейрон. Нейроны в состоянии секретировать целый набор про- и противовоспалительных цитокинов, хемокинов и факторов роста: интерлейкин-3 (IL-3), TNF-?, CXCL9 (CX-хемокиновый лиганд 9), VEGF (фактор роста эндотелия сосудов), L-селектин, IL-4, GM-CSF (гранулоцитарно-моноцитарный колониестимулирующий фактор), IL-10, IL-1R?, MIP (макрофагальный воспалительный белок) и CCL20 (C-хемокиновый лиганд 20).
Механическое растяжение изолированной культуры нейронов способствует высвобождению АТФ, который стимулирует P2X7-рецептор на собственной мембране, в результате чего нейрон секретирует цитокины. В большей мере секретируется IL-3. Интересно и то, что эти же нейроны имеют на себе рецепторы к IL-3. Предполагается, что в данном случае IL-3 обладает нейропротекторными свойствами (рис. 14). Помимо исследований in vitro, секреция IL-3 ганглионарной клеткой сетчатки при повышении давления внутри глаза подтверждается исследованиями in vivo (на мышах) [31]..
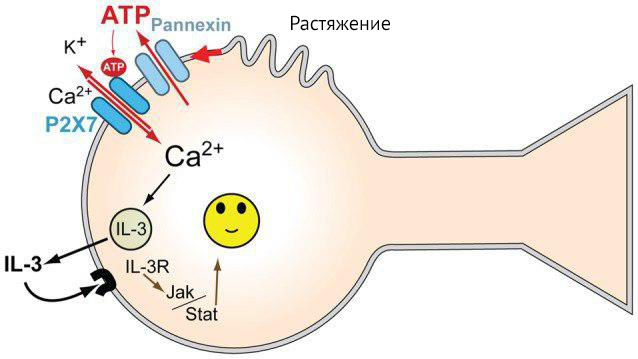
Рисунок 14 | Механизм реализации нейропротекторной функции IL-3 [31]
Основные звенья нейровоспаления открываются при изучении большого депрессивного расстройства (БДР). Множество исследований указывает на повышенные уровни воспалительных маркеров, наблюдающиеся у больных БДР. Среди них — IL-1?, IL-6, IL-18, TNF-?, IFN-?, С-реактивный белок (СРБ).
Как известно, воспаление играет немаловажную роль в микроглиальной альтерации и нарушении функций центральной серотониновой системы. Определенное воздействие оказывается и на ГГН (гипоталамо-гипофизарно-надпочечниковую ось) (рис. 15). Воспалительные цитокины повышают концентрацию кортикотропин-рилизинг гормона (КРГ), адренокортикотропного гормона (АКТГ) и кортизола и обусловливают десенситизацию глюкокортикоидных рецепторов через p38 MAPK, также приводя к нарушению работы нейромедиаторных систем, в частности норадренергической, глутаматергической и серотониновой. Вдобавок TNF-? повышает экспрессию серотонинового переносчика SERT, снижая концентрацию нейромедиатора в синаптической щели.
О стрессовом влиянии глюкокортикоидов на мозг можно прочитать в этой статье..
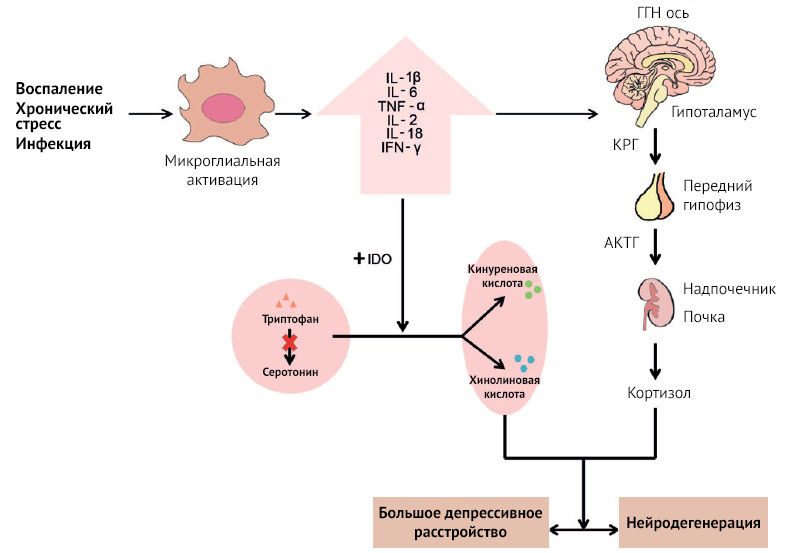
Рисунок 15
Воспаление, инфекция и стресс могут стать триггером активации микроглии. Активированная микроглия продуцирует провоспалительные цитокины, что может привести к нейродегенерации и депрессивным расстройствам через гиперактивацию ГГН оси и повышение IDO-2,3-диоксигеназной активности. Гиперактивация ГГН оси приводит к возрастанию секреции КРГ, АКТГ и кортизола, нарушая работу нейромедиаторных систем (в большей степени норадренергической и серотонинергической) и образование нейрональных факторов роста. IDO подавляет образование серотонина, смещая баланс в сторону образования KYNA и QUIN. Снижение концентрации серотонина приводит к появлению симптомов депрессии. QUIN действует как нейротоксин, глиотоксин, провоспалительный медиатор и может повреждать целостность ГЭБ [32].
Хотя активация IDO-2,3 обусловлена воспалительными цитокинами, а производные кинуренина (QUIN, KYNA) приводят к провоспалительным эффектам, IDO-2,3 является противовоспалительным медиатором. К примеру, блокаторы IDO-2,3-диоксигеназы используются в комбинации с ингибитором PD-1 в иммунотерапии опухолей (препарат Индоксимод) [34].
Основные эффекты IDO-2,3 оказывает через антигенпрезентирующие клетки (АПК): макрофаги, дендритные клетки [35] и т. д. KYN является агонистом AhR (арил-гидрокарбонового рецептора) — лиганд-активирующегося транскрипционного фактора (цитозольный сенсор ксенобиотиков), участвующего в регуляции циркадианных биоритмов, который в зависимости от лиганда проявляет разные, часто противоречащие друг другу эффекты. Активация данного рецептора может привести к дифференцировке в сторону Т-хелпера 17 (Тх17), либо Т-регуляторной клетки, что зависит от среды, а также от длительности активации. Так, кратковременная активация AhR может привести к формированию провоспалительного Тх17-лимфоцита, секретирующего IL-21, 22, 17, которые приводят к усилению воспаления, формируя положительную обратную связь. А вот длительная стимуляция AhR оказывает негативное влияние на HIF-1?, подавляет образование Тх17 и смещает дифференцировку в сторону противовоспалительных Т-регуляторных лимфоцитов (рис. 16) [36]. Под воздействием кинуренина активируется дифференциация Foxp3+ (кластер дифференцировки, ответственный за образование Т-регуляторных лимфоцитов — Tregs). Кроме того, AhR может подавлять экспрессию Nf-kB, снижая образование провоспалительных цитокинов. Активация AhR приводит к экспрессии CD39 и CD73, которые деградируют внеклеточный АТФ до противовоспалительного аденозина, о чем речь пойдет чуть позже. К противовоспалительным свойствам прибавляется также экспрессия рецепторов TIGIT (прочитать о строении и свойствах TIGIT можно здесь).
Естественно, если есть возможность так широко манипулировать иммунными клетками, обязательно найдется тот, кто воспользуется этим. Так, клетки глиобластом экспрессируют TDO (триптофан-2,3-диоксигеназу), которая метаболизирует триптофан в кинуренин (о чем мы уже говорили ранее). Кинуренин аутокринно действует на опухолевые клетки, активируя AhR и повышая пролиферацию. AhR в том числе контролирует экспрессию TGF?1, TGF?2 и латентного TGF?1-связывающего белка 1 (LAP), обусловливая рост инвазивной способности опухоли и модулируя TGF?-SMAD-путь и экспрессию интегринов (?3 и ?5) в опухолевых клетках. Более того, AhR стимулирует образование IL-6, повышая продукцию TDO и запуская положительную обратную связь. AhR оказывает влияние и на микроглию, повышая экспрессию Foxp3+ Treg, секретирующих IL-10, CD39, CD73, TIGIT, и формируя тем самым опухолевое окружение, которое приводит к росту опухоли и подавлению иммунной системы.
Стоит отметить, что хотя Тх17 играет важную роль в развитии множества аутоиммунных заболеваний, являясь источником как деструктивных, так и пролиферативных компонентов (активация Тх17 возможна при стимуляции клетки-предшественника IL-6 и трансформирующим фактором роста, который может быть причиной пролиферативных изменений, кроме того, сам Тх17 в состоянии продуцировать TGF?, что еще более усугубляет данный процесс), сам по себе Тх17 не несет в себе перманентной агрессии. Патологической Тh17 клетка становится при экспрессии рецептора к IL-23 и CC-хемокинового рецептора 6 (CCR-6). Дело в том, что стимуляция IL-21 Тх17-клетки может приводить к гиперэкспрессии IL-23R и связыванию его с IL-23, что приводит к росту устойчивости клетки через повышение экспрессии ROR?T (орфанные рецепторы, родственные рецепторам ретиноевой кислоты), IL-17, CCR-6, а также подавление IL-10. К слову, патологические Тh17 принимают участие в запоминании. Речь, правда, идет не о зубрежке, а о выученной беспомощности: Тh17, экспрессирующий CCR-6 и IL-23R, инфильтрирует гиппокамп и префронтальную кору после реализации протокола формирования беспомощности через разряды электрического тока с определенными интервалами. Использование трансгенных мышей и антител к цитокинам, определяющим дифференцировку CD4+ клеток в сторону, отличающуюся от пути дифференцировки Тх17, позволили определить главенствующую роль Тх17-клеток в формировании и прогрессировании выученной беспомощности и депрессии [37].
Если говорить о роли Т-клеточного иммунитета в ЦНС, нельзя не упомянуть, что Т-клетки в норме встречаются в цереброспинальной жидкости, периваскулярно и (в редких случаях) даже в самой паренхиме мозга. Демонстрируя надзорную функцию, Т-хелперы, не найдя мишени для активации, уходят через менингеальные лимфатические сосуды в глубокие лимфатические сосуды шеи и далее. Надзорная функция предохраняет ЦНС от оппортунистических инфекций, таких как токсоплазмоз, вирус герпеса, JC-вирус.
Регуляторные Т-клетки также выполняют надзорную роль в ЦНС, однако предохраняют они не от оппортунистических инфекций, а от избыточного иммунного ответа. Кроме того, секреция регуляторными Т-клетками BDNF, IL-10 и IL-4 демонстрирует их нейропротекторную роль при ЧМТ и нейродегенеративных заболеваниях (например, болезни Паркинсона и боковом амиотрофическом склерозе) [38]..
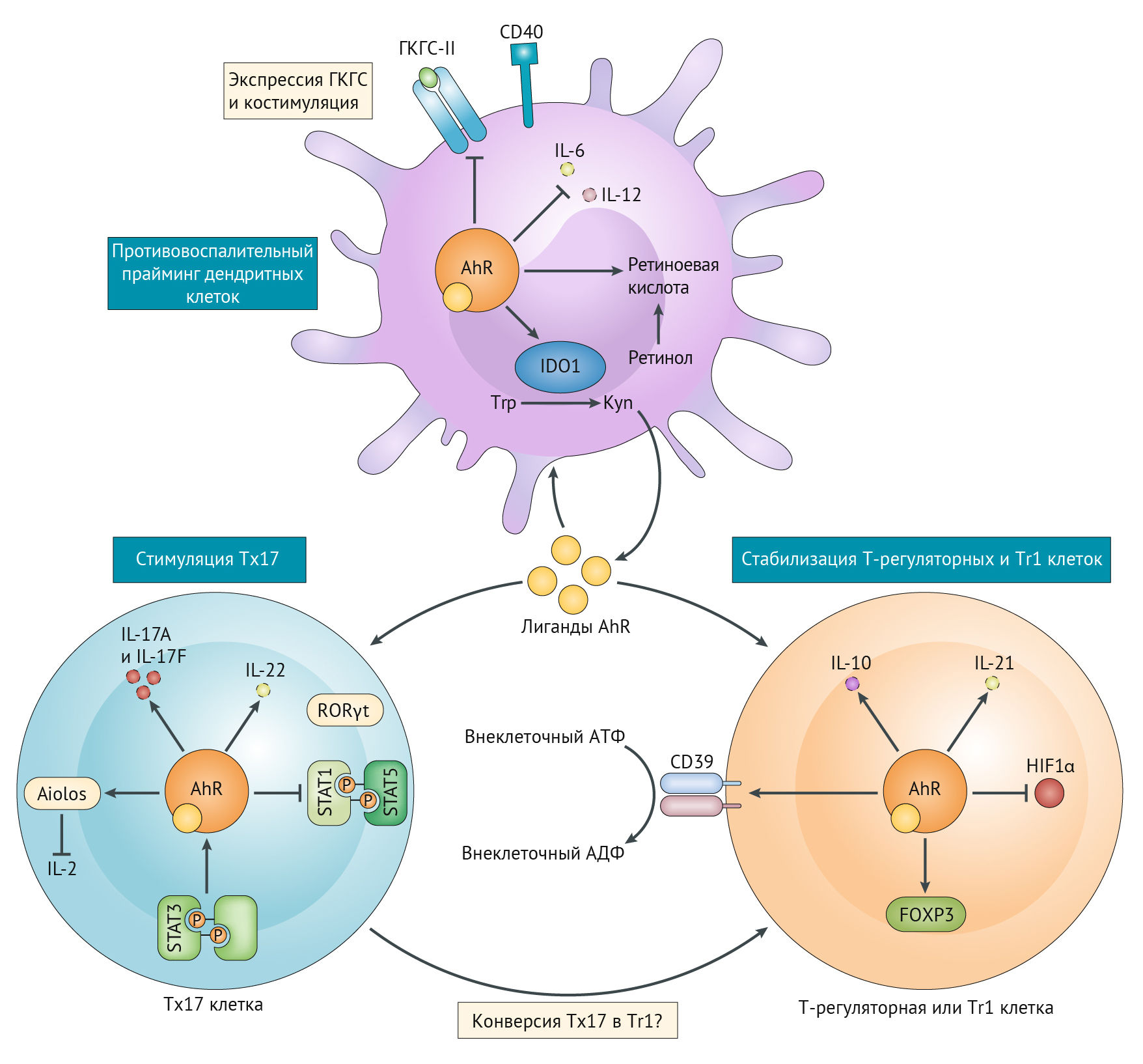
Рисунок 16
AhR имеет разный характер воздействия на дендритные клетки и на Т-клетки. В рамках провоспалительной активации в ответ на продукты обмена арахидоновой кислоты, которые также являются лигандами AhR, либо по лиганд-независимому пути активации (например, через оксидативный стресс) AhR приводит к дифференцировке Т-клетки по пути Тх17 через секрецию IL-6, ROR?T, активацию STAT3 и блокирование STAT1 и STAT5. Кроме того, важным фактором, обусловливающим дифференцировку Тх17, является индуцированный гипоксией фактор HIF-1? (несмотря на название, модулирует он дифференцировку как в состоянии гипоксии, так и в нормоксии). HIF-1? связывается с Foxp3+ и приводит к его протеасомной деградации [39]. Однако стоит понимать, что AhR и HIF-1? часто конкурируют друг с другом за ARNT (ядерный транслокатор AhR, который и реализует функцию экспрессии определенных генов, таких как IDO и TDO, а также некоторых других). Кроме связывания с ядерным аппаратом клетки, AhR играет роль E3-лигазы в убиквитин-лигазной системе деградации некоторых транскрипционных факторов, в том числе HIF-1?. При длительной стимуляции AhR наблюдается обратный эффект: активация FOXP3+, повышение TGF?, образование T-регуляторных клеток, секреция противовоспалительных цитокинов IL-10, экспрессия CD39 и CD73. Можно сказать, что AhR является фактором гомеостаза [49, 36].
Основными лигандами AhR являются продукты метаболизма кишечного микробиома (из триптофана, поступающего с пищей). AhR также участвует в поддержании целостности кишечного барьера (имеется в виду поддержание состоятельности плотных контактов между клетками эпителия кишечника) и регуляции противоопухолевого и противомикробного иммунитета в слизистой кишечника (рис. 17)..
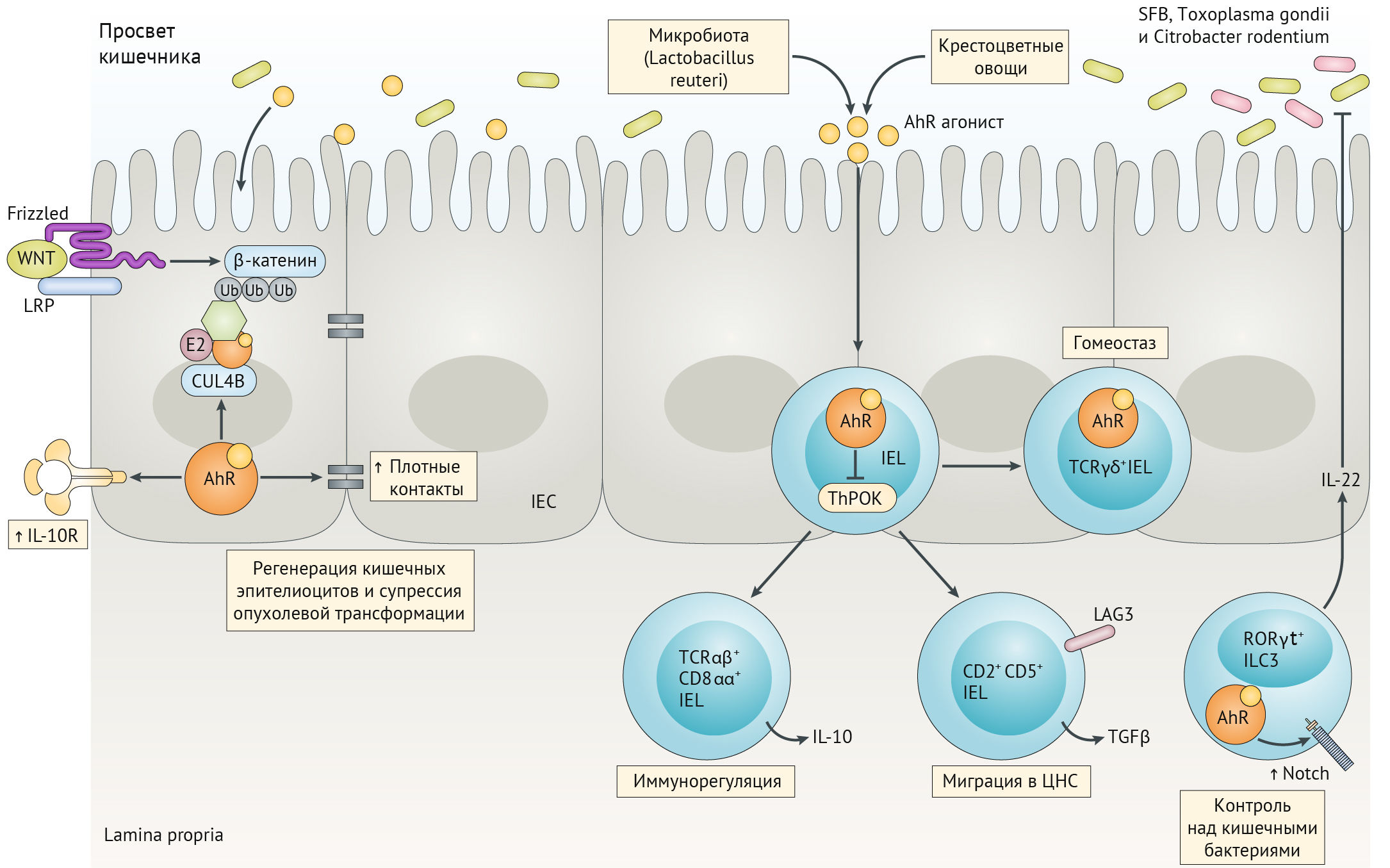
Рисунок 17
AhR модулирует регенерацию кишечных эпителиальных клеток (КЭК), предотвращая злокачественный рост. AhR опосредует противовоспалительные эффекты путем генерации интраэпителиальных лимфоцитов (IELs) и модулирует поляризацию врожденных лимфоидных клеток (ILC) для борьбы с внутрипросветными патогенами, такими как Citrobacter rodentium [36].
AhR обеспечивает нейропротокцию в ЦНС, поддерживая взаимодействие между микроглией и астроцитами (рис. 18).
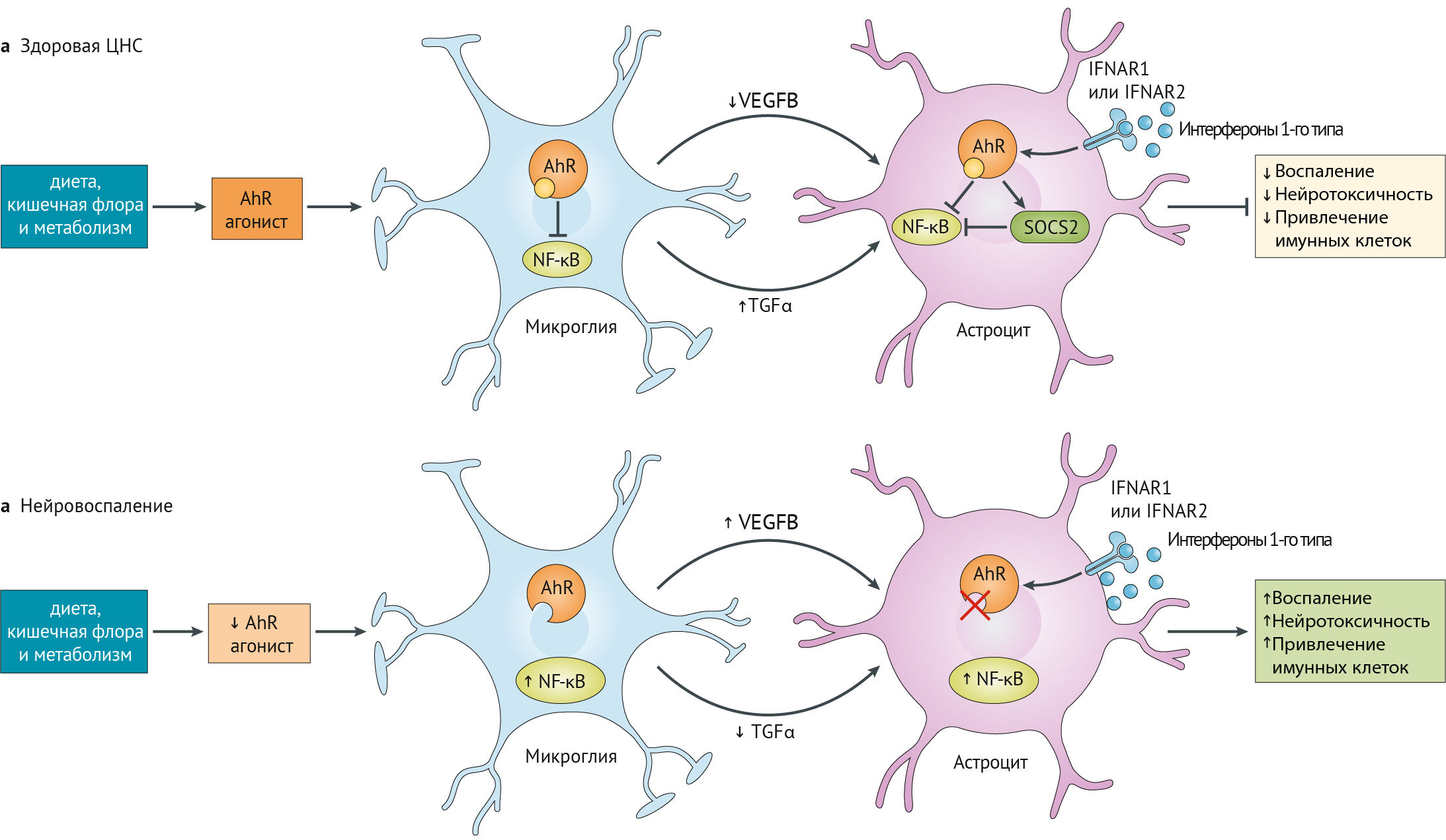
Рисунок 18
Влияние AhR на ЦНС. Нормальная деятельность AhR обеспечивает противовоспалительную функцию глии через секрецию TGF?, снижение секреции VEGF? и активацию SOCS (супрессор цитокинового сигналинга), который ингибирует NF-kB [49, 36].
В ходе метаболизма триптофана снижается сигналинг на некоторые аминокислотные сенсорные киназы, такие как GCN2 и mTOR. Отсутствие аминокислот активирует GCN2-киназу, которая фосфорилирует эукариотический инициализирующий фактор (eIF2?), подавляя рибосомальную трансляцию с большинства мРНК, но селективно повышая трансляцию с некоторых других. В CD4+ T-клетках активация GCN2 блокирует дифференцировку Тх17, способствуя созреванию Tregs. Аминокислотная депривация активирует AMPK (5'-adenosine monophosphate-activated protein kinase — протеинкиназа, активированная АМФ), которая фосфорилирует комплекс TSC1/TSC2 (комплекс туберозного склероза гамартин/туберин), что в свою очередь подавляет mTOR-сигналинг, снижая пролиферативные возможности эффекторных T-лимфоцитов [32]..
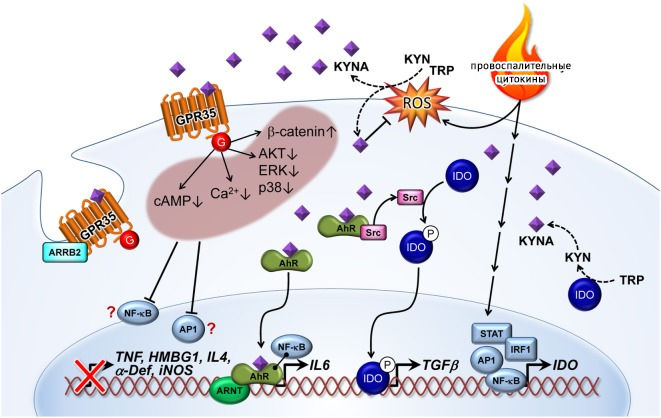
Рисунок 19
Провоспалительные цитокины индуцируют экспрессию IDO через факторы активации транскрипции, такие как STAT, AP1, IRF1 и NF-kB. KYNA формируется IDO-зависимым путем, либо альтернативным способом через трансформацию активными формами кислорода, KYN или триптофаном. С другой стороны, KYNA, являясь свободнорадикальным скавенджером, снижает уровень ROS.
KYNA связывается с рецепторами, сопряженными с g-белками 35 (GPСR35), и активирует их, снижая внутриклеточные уровни цАМФ и Ca2+. GPCR35 экспрессируются на множестве субпопуляций иммунных клеток, включая периферические моноциты, мастоциты, базофилы, эозинофилы и iNKT-клетки (invariant natural killer T-cells). KYNA-GPСR35-опосредованное ингибирование аденилатциклазы приводит к снижению активности IL-23/IL-17 иммунной оси. Активация GPCR35 кинурениновой кислотой может как ингибировать фосфорилирование протеинкиназы B (AKT), ERK (киназа, регулируемая внеклеточными сигналами) и p38 (p38 митоген-активируемая протеинкиназа), так и повышать уровень ?-катенина. Все эти клеточные взаимодействия, вероятно, снижают активацию соответствующих воспалительных факторов транскрипции, таких как NF-?B и AP1. Таким образом, снижение активации TNF-?, амфотерина (high-mobility group box 1, HMBG1), IL-4, ?-дефензина (?-def) и индуцибельной синтазы оксида азота (iNOS) часто наблюдалось в ответ на повышение концентрации KYNA.
Аррестин-B2 (ARRB2) в GPCR35 необходимы для интернализации и десенситизации активированного KYNA рецептора.
Связывание KYNA с рецептором AhR приводит к активации ARNT и индукции экспрессии IL-6. Взаимодействие комплекса KYNA-AhR с NF-?B также может быть вовлечено в экспрессию IL-6. Кроме того, активированный лигандом AhR инициирует активацию протоонкогена тирозин-протеинкиназы Src и далее — фосфорилирование IDO. Фосфорилированный IDO индуцирует экспрессию трансформирующего фактора роста ?1 (TGF?1) [7].
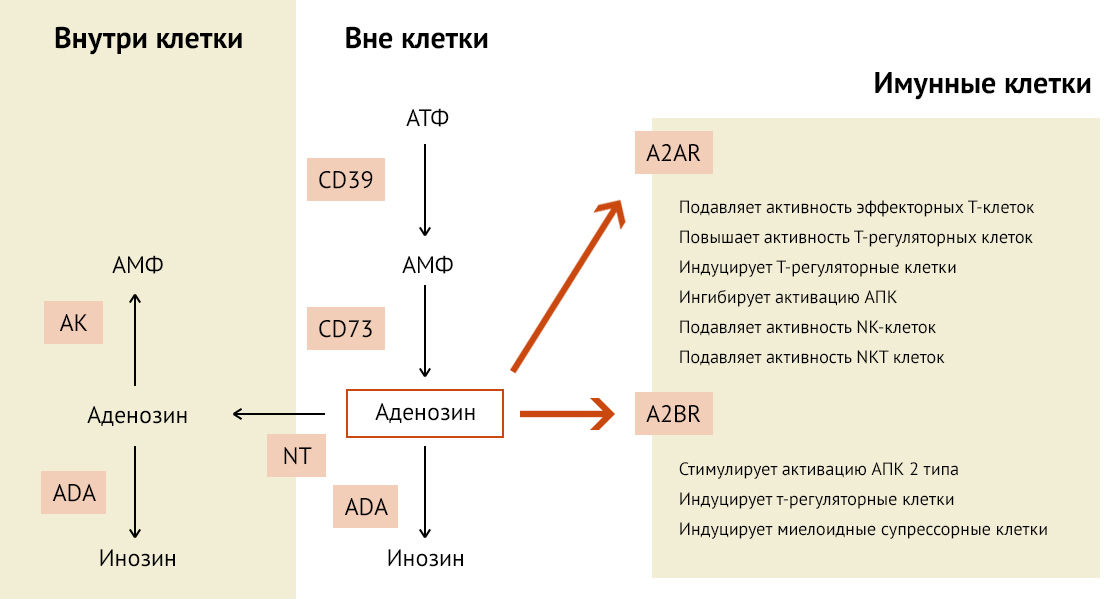
Рисунок 20
CD39 и CD73 продуцируют внеклеточный аденозин. Концентрация внеклеточного аденозина снижается с помощью аденозиндезаминаза (ADA)-зависимого катаболизма и путем его поглощения клетками через нуклеотидные транспортеры (NT). Аденозин во внутриклеточном компартменте преобразуется в АМФ с помощью аденозинкиназы (AK) или катаболизируется с помощью ADA. При повышении внеклеточного уровня аденозина он стимулирует A2AR (высокоаффинные) и A2BR (низкоаффинные) рецепторы на иммунных клетках. Аденозин проявляет супрессивные свойства для эффекторных клеток T (Teff), NK и NKT. Иммунодепрессивная активность может быть дополнительно усилена аденозин-опосредованной индукцией Tregs, толерогенными АПК (антиген-презентирующие клетки) и MDSC (Myeloid-derived suppressor cells — супрессорные клетки миелоидного происхождения, включают незрелые формы макрофагов, гранулоцитов, дендритных клеток, экспрессируют иммуносупрессивные молекулы (аргиназу и активные формы кислорода/азота)) и особенно угнетением активности T-клеток [40].
CD39 и CD73 участвуют в сохранении ветвистой формы микроглии, объем которой способствует поддержанию стабильного иммунологического состояния ЦНС [41]. В данном случае аденозин препятствует дифференцировке надзорной микроглиальной в М1-активированную провоспалительную клетку. Так как АТФ является алармином и запускает иммунный ответ, способствуя формированию M1-фенотипа микроглии, то его метаболит (аденозин) в случае классической активации микроглии способствует смещению поляризации в сторону M2-фенотипа [42].
.
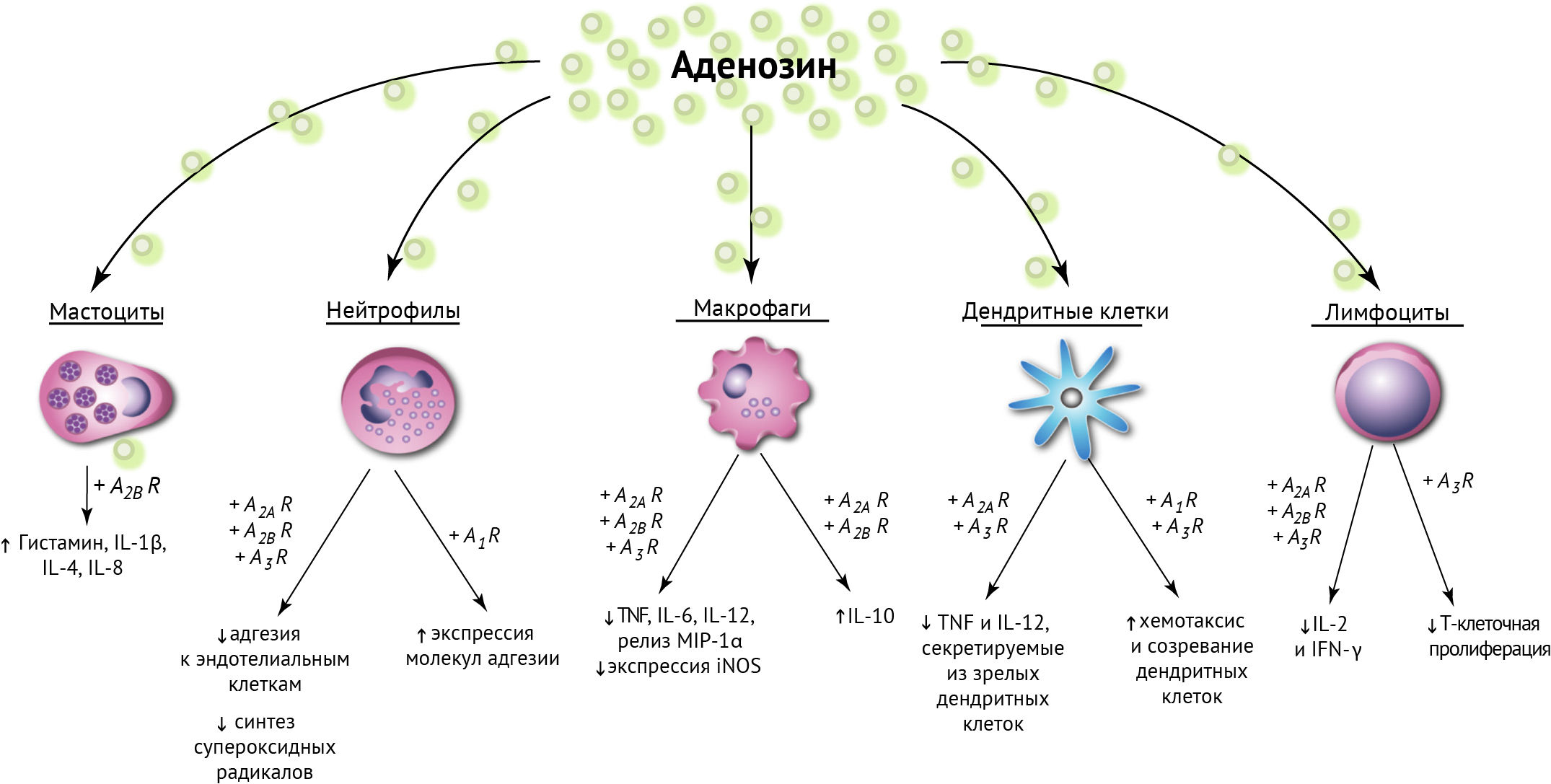
Рисунок 21
Аденозин распространяет свое влияние на большинство иммунокомпетентных клеток, подавляя их функцию либо смещая в сторону противовоспалительной полярности [42].
И хотя аденозин имеет иммуносупрессивные свойства, отсутствие CD73 в экспериментальном аутоиммунном энцефаломиелите не оказало никакого эффекта ни на биохимическую составляющую (не снижало экспрессию Тх17, а повышение экспрессии на их поверхности CD73 не влияло на пролиферацию и созревание Treg), ни на симптоматику самого заболевания [43].
A2BR-активация имеет спорное значение. К примеру, применение антагониста A2BR на модели экспериментального аутоиммунного энцефаломиелита (особенно подавление образования IL-6 и дифференцировки Тх17) продемонстрировало значительное снижение воспалительной активности [44].
Развитие нервной системы
Большинство исследований все же ретроспективны, и причинно-следственную связь обнаружить непросто. Трудно обозначить, является ли гиперактивность иммунной системы причиной, либо же она просто сопутствует основному повреждающему фактору. Какое влияние оказывает антенатальное и постнатальное воспаление?
Клетки микроглии выполняют функции не только иммунокомпетентных клеток. Кроме активации апоптоза и фагоцитоза, секреции цитокинов в ответ на повреждающий фактор, они также вовлечены в аксональное развитие, синаптогенез и синаптический прунинг (процесс, обратный синаптогенезу, заключающийся в снижении количества межсинаптических связей). Принимая во внимание важность микроглиальной активности, стоит отметить, что без активной поддержки микроглии клетки пятого слоя коры головного мозга не развиваются и впоследствии подвергаются апоптозу в постнатальном периоде. CX3CR1 в микроглии необходим для выживания пятого слоя коры мозга. Микроглиальный трофический фактор роста IGF-1 предотвращает наступление апоптоза нейронов. Новорожденные мыши, подвергнутые неинфекционному нейровоспалению, на примере гипоксии/ишемии головного мозга имели повышенные уровни глиального фибриллярного кислого белка (GFAP) — маркера астроглиоза.
Трансгенные мыши с отсутствием GFAP демонстрировали пониженный реактивный глиоз после гипоксии/ишемии, однако объем поражения мозга не был снижен. И все же относительно контрольной группы эти мыши имели большее количество выживших нейронов в зубчатой извилине гиппокампа. С другой стороны, астроциты продуцируют цитокины, например, лейкемия-ингибирующий фактор (LIF), стимулирующий регенеративный ответ нейрональных стволовых клеток в субвентрикулярной зоне мозга после неонатальной гипоксии/ишемии [45]. Ранняя активация микроглии может играть роль в развитии расстройств аутистического спектра (РАС). В исследованиях описывается как нарушение прунинга и увеличение общего количества дендрит-дендритных связей в определенных областях мозга, так и отсутствие такой закономерности, что не исключает попыток рассматривать данный признак как одну из возможных причин развития РАС, но в то же время не дает возможности назвать его единственным этиологическим фактором. Кроме того, существует гипотеза о синаптической дезорганизации и дисфункции, сторонники которой отмечают дефицит глиальных факторов, развившийся, вероятно, в результате воспаления [46]. В дополнение к факту влияния нейровоспаления и микроглиальной альтерации на развитие психических заболеваний можно привести в пример исследования, в которых отмечается связь между психическими заболеваниями пациентов и воспалительным процессом у их матерей во время беременности [47]. Активация TLR-3 путем имитации вирусной экспансии через введение в организм синтетической двухцепочечной РНК — полиинозин-полицитидиловой кислоты (poly I:C, структурно похож на двуцепочечную РНК, которая содержится у некоторых вирусов) применяется в качестве мышиной модели шизофрении.
Использование миноциклина либо глубокая стимуляция головного мозга новорожденных крыс может предотвратить поведенческий и структурный дефицит в старшем возрасте.
Пренатальная экспозиция аллергенов играет роль в формировании тех или иных паттернов социального и полового поведения в будущем.
В недавнем исследовании Lenz et al (2019) продемонстрировано влияние повышенной активности микроглии и мастоцитов в преоптической области мозга в ответ на внутриутробную экспозицию аллергенов. Так, кроме активации микроглии было заметно увеличено количество дендритных шипиков у нейронов преоптической области мозга, что, по всей вероятности, отвечает за маскулинное поведение. Среди мышей, подвергнутых внутриутробно экспозиции аллергенами, во взрослом периоде даже для женских особей было характерно маскулинное поведение. Данное явление, возможно, связано с вмешательством в обмен нейростероидов в критический период внутриутробного развития. Разрастание дендритных шипиков объясняется повышенной секрецией PGE2 (простагландина E2) активированной микроглией. Модель аллергического воспаления объясняет активацию микроглии через выделение гистамина мастоцитами [48].
Мастоциты также вовлечены в активное взаимодействие с ЦНС и ПНС. В частности, иммуномодулирующий эффект, наблюдающийся при активации мастоцитов (учитывая расположение их в области твердой и мягкой мозговых оболочек в непосредственной близости к задним корешковым ганглиям), приводит к повышению проницаемости ГЭБ, развитию нейропатических болей, централизации периферической боли и т. д. (рис. 20). Интересны механизмы, при помощи которых мастоциты «общаются» с глией и нейронами — через передачу внеклеточных везикул. Из-за своего маленького размера везикулы способны преодолевать большие расстояния и влиять на нейротрансмиссию в ЦНС [49]..
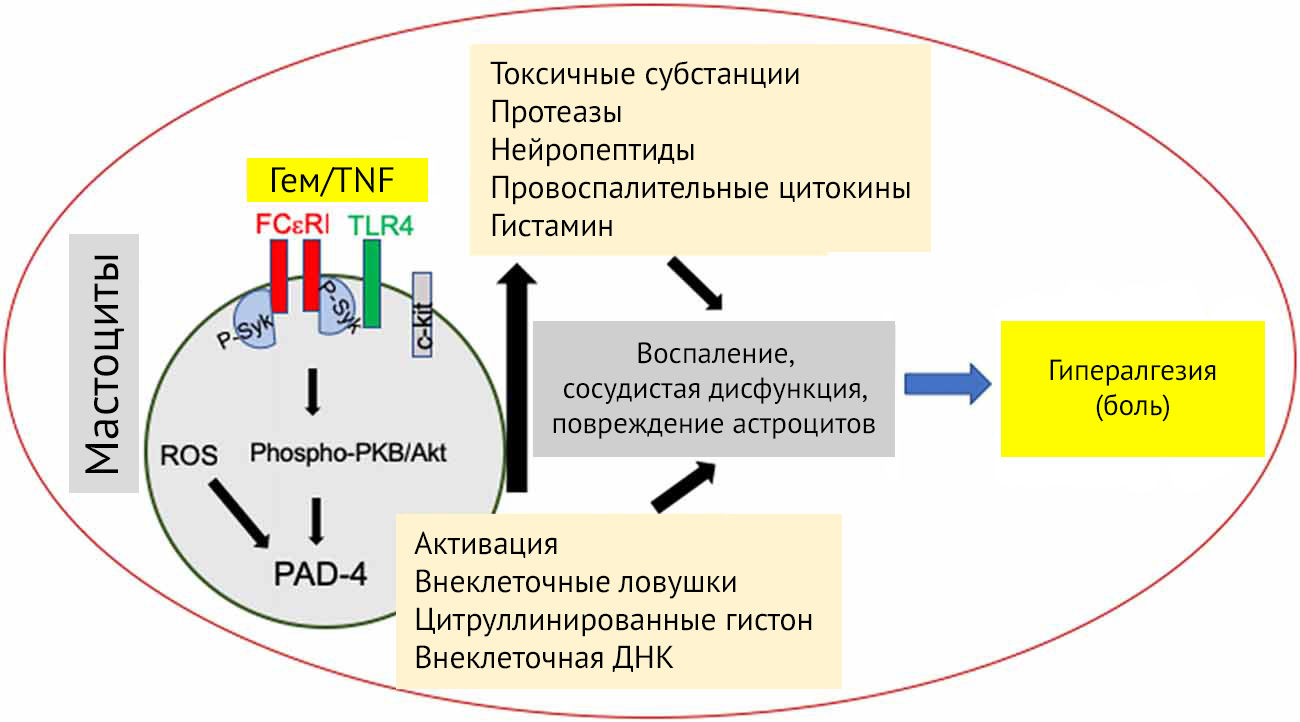
Рисунок 22
Механизм активации мастоцита и последующая дегрануляция, образование внеклеточных везикул и мастоцитарных ловушек, оказывающих негативное влияние на функцию ГЭБ и астроцитов в частности.
Дегрануляция начинается с активации рецепторов, высокоаффинных к IgE (Fc?R1), перегруппировки F-актина и образования микротрубочек. Fc?R1 активирует Fyn/Gab2/RhoA-тирозинкиназы, приводит к полимеризации микротрубочек и перемещению секреторных гранул к цитоплазматической мембране. Тирозинкиназы Lyn и Syk, будучи активированными через Fc?R1, вызывают мобилизацию кальция из внутриклеточных компартментов, слияние везикул и экзоцитоз гранул через активацию пути протеинкиназы B и апрегуляции пептидил-аргинин дезаминазы-4 (PAD-4) [49].
Кроме того, активацию мастоцитов рассматривают в качестве одного из возможных звеньев патогенеза болезни Паркинсона: мастоциты способствуют активации NF-kB в астроцитах, микроглии, а также повышению секреции этими клетками IL-33 — цитокина, концентрация которого заметно выше у пациентов с болезнью Паркинсона [50].
Гуморальные факторы, такие как система комплемента, принимают активное участие в пренатальном и постнатальном развитии мозга. Критически важны белки системы комплемента во время нейруляции — миграции клеток будущей ЦНС. В постнатальном периоде C1q и C3 участвуют в синаптическом прунинге, их дефицит приводит к улучшению памяти и гиппокамп-зависимому обучению. Однако избыточное количество межсинаптических связей повышает риск развития эпилепсии [17].
В развитии ЦНС (да и в целом всего организма) немаловажную роль играют хемокины. Однако только CXCL12/CXCR4/ACKR3 являются критически важными для выживания организма. Делеция генов, обусловливающих их экспрессию, грозит внутриутробной гибелью плода.
На схеме далее (рис. 23) проиллюстрированы функции хемокинов, изученные на данный момент [51]..

Рисунок 23 | Функции хемокинов
Микроглия также играет заметную роль в нейропластичности, дыхании, ноцицепции [52], совместно с астроцитами влияет на сон [53].
Микроглиальные клетки отвечают за формирование, развитие и защиту мозга, вовлечены в реализацию физиологических процессов. Противоречивые данные о наличии воспаления, сопровождающего отдельные психические или неврологические заболевания, могут навести на мысль, что дисфункция микроглии и дизрегуляция квадрисинаптической системы играет более важную роль, чем развитие воспаления, которое может являться лишь следствием (имеется в виду хроническое воспаление, а не острое, когда основной вклад в повреждение ЦНС вкладывает патоген и агрессия иммунной системы).
Терапия
Антидепрессанты: в исследовании на животных Obuchowicz et al (2014) после введения подопытным LPS применение имипрамина и флуоксетина привело к снижению секреции IL-6, IL-1?, TNF-? микроглией, но только имипрамин предотвращал морфологические изменения и активацию микроглии. В другом исследовании (Xia et al, 1996) имипрамин, кломипрамин и циталопрам оказывали схожий эффект. Кломипрамин, сертралин и тразодон снижали уровень IFN-?, повышали продукцию противовоспалительного цитокина IL-10 [32].
В целом, исследования на пациентах подтверждают корреляцию между приемом антидепрессантов и противовоспалительными эффектами (табл. 3).
Таблица 3 | Связь между приемом антидепрессантов и уровнем провоспалительных цитокинов [32]
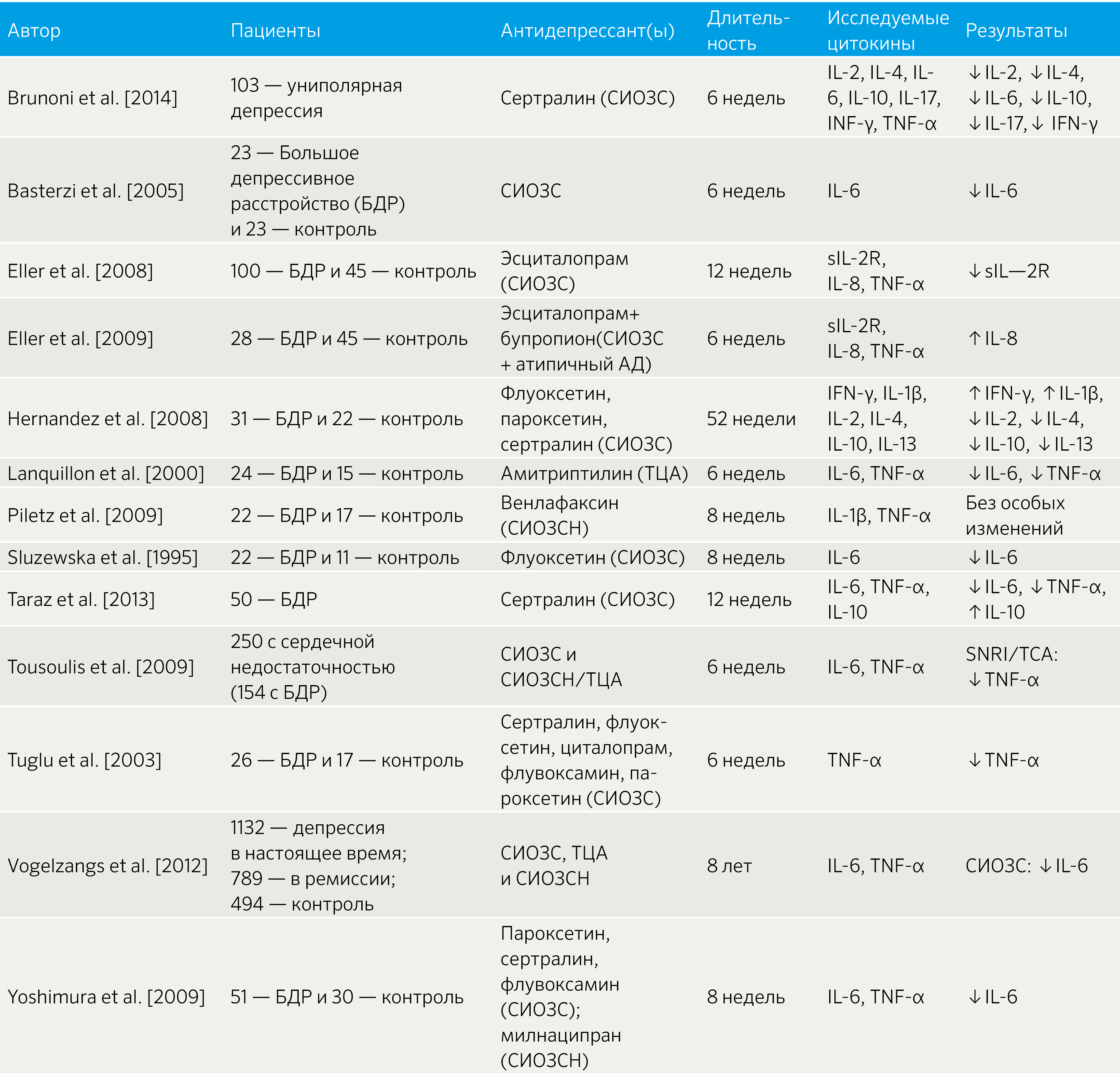
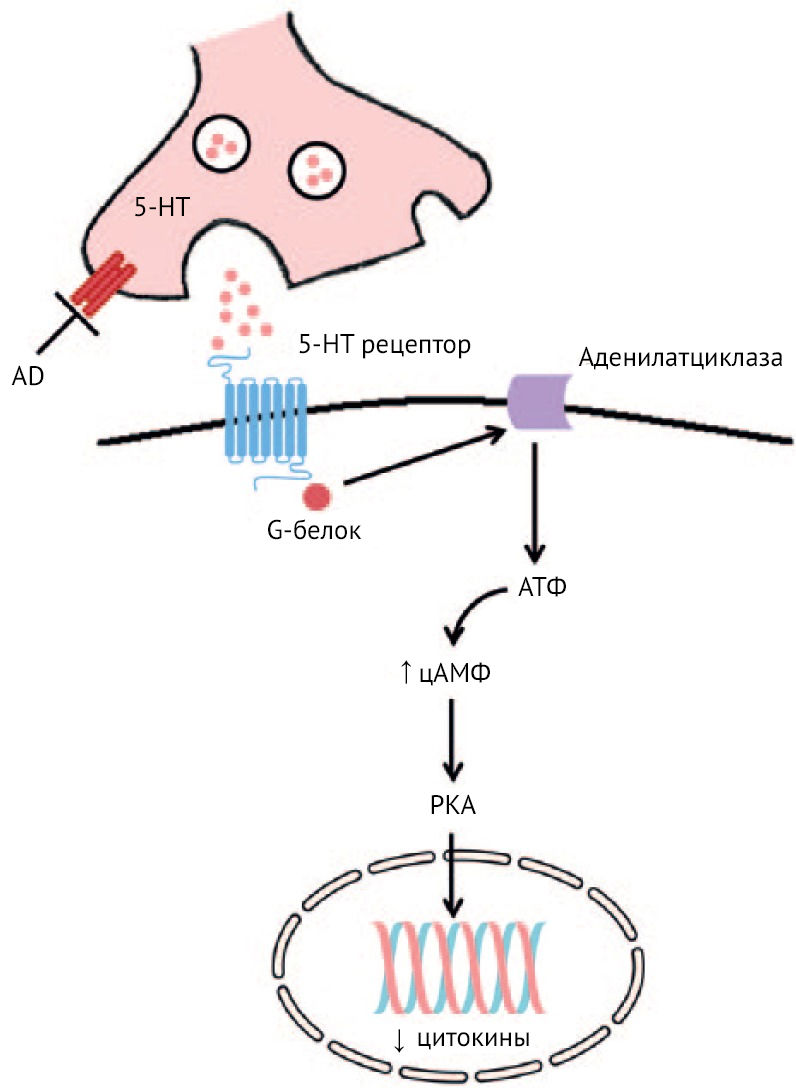
Рисунок 24 | Механизм противовоспалительного действия антидепрессантов
Примечательно, что противовоспалительный эффект связывают с повышением активности аденилатциклазы в моноцитах и лимфоцитах. Повышение уровня цАМФ связывают со снижением фагоцитирующей и киллерной функций и уменьшением секреции воспалительных цитокинов (рис. 24) [31, 54]. Но это несколько противоречит описанному ранее снижению активации аденилатциклазы через связывание KYNA с GPCR35 и, как следствие, снижению провоспалительной активности иммунных клеток. Данный эффект связан с консервативным сигнальным путем Notch. Интересно, что, способствуя активации Notch совместно с активацией TLR, протеинкиназа А (PKA) приводит к формированию сильной отрицательной обратной связи в ответ на секрецию IL-6, IL-12, IFN? через репрессорный транскрипторный комплекс Groucho/трансдуциноподобный энхансер Split (TLE) и Hes1 (ген Notch), что в итоге приводит к вышеописанным явлениям.
НПВС: Ингибиторы циклооксигеназы (ЦОГ) используются в терапии некоторых психических расстройств, часто в комбинации с антидепрессантами. В качестве блокатора циклооксигеназы хорошее впечатление о себе оставляет целекоксиб — селективный ингибитор ЦОГ-2. При комбинировании с антидепрессантами он показал наилучший эффект относительно плацебо [55, 56].
Терапия НПВС приводила к улучшению состояния пациентов с шизофренией при условии раннего начала терапии. Сочетание рисперидона с целекоксибом демонстрирует лучшие показатели, чем рисперидона с плацебо. Тем не менее при длительном течении шизофрении комбинированная терапия не демонстрирует выраженного эффекта [57].
Ингибиторы цитокинов (например, моноклональные антитела к TNF-? — инфликсимаб) снижали депрессивную симптоматику у пациентов с псориазом. Хотя у пациентов, невосприимчивых к лечению данным препаратом, депрессивная симптоматика осталась без изменений.
Вагус-стимуляция
Противовоспалительная активность реализуется несколькими путями:
- Воздействие на ГГН ось через афферентные окончания, что приводит к секреции кортизола надпочечниками.
- Холинергический путь — высвобождение эфферентами ацетилхолина, который воздействует на ?7NAChR на поверхности макрофага, что приводит к ингибированию синтеза воспалительных цитокинов, таких как TNF-?.
- Стимуляция симпатических волокон НС селезенки, высвобождение норадреналина, связывание с ?2-адренергическими рецепторами на селезеночных лимфоцитах, которые высвобождают ацетилхолин, далее — по холинергическому пути.
Данный метод нашел применение в терапии эпилепсии и психических заболеваний, таких как униполярная и биполярная депрессии [58]. Кроме того, неинвазивная вагус-стимуляция уменьшает проницаемость ГЭБ и объем поражения мозга у мышей с транзиторной окклюзией средней мозговой артерии [59].
Глубокая стимуляция мозга
Согласно упоминавшемуся ранее исследованию [47], глубокая стимуляция мозга новорожденных мышей, матери которых были заражены инфекцией в антенатальный период, предотвращала микроглиальную альтерацию. Глубокая стимуляция мозга у мышей с индуцированной депрессией вызывает противовоспалительный и антиапоптотический эффекты [60]. Тем не менее у пациентов с болезнью Паркинсона и болезнью Альцгеймера глубокая стимуляция мозга повышает уровень про-гепцидина в мозге, что, вероятно, может приводить к ферроптозу [61].
Ранее уже приводились примеры, в контексте которых описывались положительные моменты нейровоспаления. В заключение стоит привести исследование, касающееся применения PD-1-ингибиторов (Programmed cell Death-1 — иммунная контрольная точка в терапии онкологических заболеваний) на мышиной модели болезни Альцгеймера (5XFAD — 5 семейных мутаций, связанных с болезнью Альцгеймера) [62]. Ингибитор PD-1 приводил к повышению уровня IFN-? у мышей, снижению концентрации Tregs, увеличению проницаемости хориоидального сплетения, миграции макрофагов и Tregs в ЦНС, очищению от ?-амилоидных бляшек, улучшению когнитивного статуса.
Увеличение концентрации IFN-? сопровождается ростом уровня CCR-2 (CC-хемокиновый рецептор-2, обладает скорее иммуномодулирующим, нежели просто провоспалительным эффектом; считается, что основная роль CCR-2 заключается в мобилизации моноцитов; помимо функции активации микроглии по M1-фенотипу, CCR-2 может обладать иммуносупрессивными свойствами в зависимости от множества обстоятельств [63]), инфильтрацией паренхимы мозга моноцитами, повышением экспрессии скавенджер-рецептора A (SRA1). Лечение приводило к улучшению когнитивных функций (тестировались пространственное обучение и память), а также к уменьшению количества ?-амилоидных бляшек в гиппокампе и V слое коры головного мозга. Была снижена выраженность астроглиоза в гиппокампе.
Источники:
- Caroline Menard, Madeline L. Pfau. at al. “Social stress induces neurovascular pathology promoting depression” Nature Neuroscience | VOL 20 | NOVEMBER 1752 2017 | 1752–1760
- Ю.Е. Шилов, М.В. Безруков “Кинуренины в патогенезе эндогенных психических заболеваний” ВЕСТНИК РАМН /2013/ № 1
- Carlos R. Dostal, Megan Carson Sulzer at al “Glial and tissue-specific regulation of Kynurenine Pathway dioxygenases by acute stress of mice” Neurobiol Stress. 2017 Dec; 7: 1–15.
- Joshua Chiappelli, MD, Ana Pocivavsek, PhD, at al “Stress-Induced Increase in Kynurenic Acid as a Potential Biomarker for Patients With Schizophrenia and Distress Intolerance” JAMA Psychiatry. 2014 Jul 1; 71(7): 761–768.
- Chiappelli J1, Postolache TT at al “Tryptophan Metabolism and White Matter Integrity in Schizophrenia” Neuropsychopharmacology. 2016 Sep;41(10):2587-95.
- Joshua Chiappelli, Laura M. Rowland, “Salivary kynurenic acid response to psychological stress: inverse relationship to cortical glutamate in schizophrenia” Neuropsychopharmacologyvolume 43, pages1706–1711 (2018)
- Elisa Wirthgen, Andreas Hoeflich, at al “Kynurenic Acid: The Janus-Faced Role of an Immunomodulatory Tryptophan Metabolite and Its Link to Pathological Conditions” Front Immunol. 2017; 8: 1957.
- Wu HQ1, Rassoulpour A, Schwarcz R. “Kynurenic acid leads, dopamine follows: a new case of volume transmission in the brain?” J Neural Transm (Vienna). 2007 Jan;114(1):33-41.
- http://psyandneuro.ru/
- R?us GZ, Becker IRT at al “The inhibition of the kynurenine pathway prevents behavioral disturbances and oxidative stress in the brain of adult rats subjected to an animal model of schizophrenia” Prog Neuropsychopharmacol Biol Psychiatry. 2018 Feb 2;81:55-63.
- Harikesh Kalonia, Jitendriya Mishra, Anil Kumar “Targeting Neuro-Inflammatory Cytokines and Oxidative Stress by Minocycline Attenuates Quinolinic-Acid-Induced Huntington’s Disease-Like Symptoms in Rats” Neurotoxicity Research November 2012, Volume 22, Issue 4, pp 310–320.
- Paula Pierozan,Helena Biasibetti at al “Quinolinic acid neurotoxicity: Differential roles of astrocytes and microglia via FGF-2-mediated signaling in redox-linked cytoskeletal changes” Biochimica et Biophysica Acta (BBA) - Molecular Cell Research Volume 1863, Issue 12, December 2016, Pages 3001-3014
- Cataldo Arcuri, Carmen Mecca at al “The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing CNS” Front Mol Neurosci. 2017; 10: 191.
- Yochai Wolf, Simon Yona, Ki-Wook Kim, and Steffen Jung. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci. 2013; 7: 26.
- Douglas G Walker and Lih-Fen Lue. Understanding the neurobiology of CD200 and the CD200 receptor: a therapeutic target for controlling inflammation in human brains? Future Neurol. 2013 May; 8(3).
- Pluvinage JV, Haney MS, Smith BAH, et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature. 2019 Apr;568(7751):187-192.
- Carpanini Sarah M., Torvell Megan, Morgan Bryan Paul. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol., 04 March 2019.
- Botto M1, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiology. 2002 Sep;205(4-5):395-406.
- Lubka T. Roumenina, Damien S?ne, Maria Radanova, et al. Functional Complement C1q Abnormality Leads to Impaired Immune Complexes and Apoptotic Cell Clearance. J Immunol October 15, 2011, 187 (8) 4369-4373.
- B. Paul Morgan. Complement in the pathogenesis of Alzheimer’s disease. Semin Immunopathol. 2018; 40(1): 113–124.
- Kristina A. Kigerl, Juan Pablo de Rivero Vaccari at al “Pattern recognition receptors and central nervous system repair” Exp Neurol. 2014 Aug; 258: 5–16.
- Jamie S. Church, Kristina A. Kigerl at al “TLR4 Deficiency Impairs Oligodendrocyte Formation in the Injured Spinal Cord” J Neurosci. 2016 Jun 8; 36(23): 6352–6364.
- Christiane Ott, Kathleen Jacobs, Elisa Haucke, Anne Navarrete Santos, Tilman Grune, Andreas Simm. Role of advanced glycation end products in cellular signaling. Redox Biology 2 (2014) 411–429.
- Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173(4):649–665.
- https://biomolecula.ru/
- Zhang L, Zhang J, You Z. Switching of the Microglial Activation Phenotype Is a Possible Treatment for Depression Disorder. Front Cell Neurosci. 2018;12:306. Published 2018 Oct 16.
- Choi MJ, Lee EJ, Park JS, Kim SN, Park EM, Kim HS. Anti-inflammatory mechanism of galangin in lipopolysaccharide-stimulated microglia: Critical role of PPAR-? signaling pathway. Biochem Pharmacol. 2017 Nov 15;144:120-131.
- Nnah IC, Wessling-Resnick M. Brain Iron Homeostasis: A Focus on Microglial Iron. Pharmaceuticals (Basel). 2018;11(4):129.
- Abigail Weiland, Yamei Wang, Weihua Wu, Xi Lan, Xiaoning Han, Qian Li, Jian Wang. Ferroptosis and Its Role in Diverse Brain Diseases. Mol Neurobiol. 2019 Jul;56(7):4880-4893.
- Veit Rothhammer and Francisco J. Quintana “Control of autoimmune CNS inflammation by astrocytes” Semin Immunopathol. 2015 Nov; 37(6): 625–638.
- Lim JC, Lu W, Beckel JM, Mitchell CH. Neuronal Release of Cytokine IL-3 Triggered by Mechanosensitive Autostimulation of the P2X7 Receptor Is Neuroprotective. Front Cell Neurosci. 2016;10:270.
- Paula Kopschina Feltes,1,2 Janine Doorduin, at al “Anti-inflammatory treatment for major depressive disorder: implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy” J Psychopharmacol. 2017 Sep; 31(9): 1149–1165.
- L. Brundin, S. Erhardt, at al “The role of inflammation in suicidal behaviour” Acta Psychiatr Scand. 2015 Sep; 132(3): 192–203.
- https://www.cancer.gov/
- David H. Munn, Andrew L. Mellor “Indoleamine 2,3 dioxygenase and metabolic control of immune responses” Trends Immunology March 2013 Volume 34, Issue 3, p137–143.
- Veit Rothhammer and Francisco J. Quintana. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol. 2019 Mar;19(3):184-197.
- El?onore Beurel, Jeffrey Lowell, Richard Jope. Distinct characteristics of hippocampal pathogenic TH17 cells in a mouse model of depression. Brain, Behavior, and Immunity. Volume 73, October 2018, Pages 180-191.
- Erik Ellwardt, JamesT.Walsh, Jonathan Kipnis, and Frauke Zipp. Understanding the role of T Cells in CNS Homeostasis. Trends Immunol. 2016 Feb;37(2):154-165.
- Dang EV, Barbi J, Yang HY, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146(5):772–784.
- Akio Ohta, Michail Sitkovsky “Extracellular Adenosine-Mediated Modulation of Regulatory T Cells” Front Immunol. 2014; 5: 304.
- Marina Matyash, Oleksandr Zabiegalov, at al “The adenosine generating enzymes CD39/CD73 control microglial processes ramification in the mouse brain” PLoS One April 4, 2017.
- Luca Antonioli, Bal?zs Cs?ka, at al “Adenosine and inflammation: what's new on the horizon?” Drug Discovery Today Volume 19, Issue 8, August 2014, Pages 1051-1068.
- Gerard Hernandez-Mir and Mandy J. McGeachy “CD73 is expressed by inflammatory Th17 cells in experimental autoimmune encephalomyelitis but does not limit differentiation or pathogenesis” PLoS One. 2017; 12(3): e0173655.
- Wei Wei, Changsheng Du, at al “Blocking A2B Adenosine Receptor Alleviates Pathogenesis of Experimental Autoimmune Encephalomyelitis via Inhibition of IL-6 Production and Th17 Differentiation” J Immunol January 1, 2013, 190 (1) 138-146.
- Luba Sominsky, Simone De Luca, Sarah J.Spencer, at al “Microglia: Key players in neurodevelopment and neuronal plasticity” The International Journal of Biochemistry & Cell Biology Volume 94, January 2018, Pages 56-60.
- Amin Mottahedin, Maryam Ardalan at al “Effect of Neuroinflammation on Synaptic Organization and Function in the Developing Brain: Implications for Neurodevelopmental and Neurodegenerative Disorders” Front Cell Neurosci. 2017; 11: 190.
- Hadar R, Dong L, at al “Deep brain stimulation during early adolescence prevents microglial alterations in a model of maternal immune activation” Brain Behav Immun. 2017 Jul;63:71-80.
- Kathryn M. Lenz, Lindsay A. Pickett, Christopher L. Wright, Anabel Galan & Margaret M. McCarthy. Prenatal Allergen Exposure Perturbs Sexual Differentiation and Programs Lifelong Changes in Adult Social and Sexual Behavior. Scientific Reports volume 9, Article number: 4837 (2019).
- Aditya Mittal, Varun Sagi, Mihir Gupta and Kalpna Gupta. Mast Cell Neural Interactions in Health and Disease.Front. Cell. Neurosci., 20 March 2019.
- Duraisamy Kempuraj, Ramasamy Thangavel et al. Mast Cell Proteases Activate Astrocytes and Glia-Neurons and Release Interleukin-33 by Activating p38 and ERK1/2 MAPKs and NF-?B. Molecular Neurobiology. 12 April 2018.
- Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. 2018;285(16):2944–2971.
- Austin D. Hocker, Jennifer A. Stokes, at al “The impact of inflammation on respiratory plasticity” Exp Neurol. 2017 Jan; 287(Pt 2): 243–253.
- Agnes Nadjar, Tamara Blutstein, at al “Astrocyte?derived adenosine modulates increased sleep pressure during inflammatory response” GLIA 2013;61:724–731.
- Jason L. Larabee, Salika M. Shakir, at al “Increased cAMP in Monocytes Augments Notch Signaling Mechanisms by Elevating RBP-J and Transducin-like Enhancer of Split (TLE)” J Biol Chem. 2013 Jul 26; 288(30): 21526–21536.
- Akhondzadeh S1, Jafari S, at al “Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial” Depress Anxiety. 2009;26(7):607-11. doi: 10.1002/da.20589.
- Faridhosseini F, Sadeghi R, Farid L, Pourgholami M “Celecoxib: a new augmentation strategy for depressive mood episodes. A systematic review and meta-analysis of randomized placebo-controlled trials” Human Psychopharmacology 2014; 29(3): 216-223.
- Norbert M?ller, MD, PhD “Immunological aspects of the treatment of depression and schizophrenia” Dialogues Clin Neurosci. 2017 Mar; 19(1): 55–63.
- John P. O'Reardon, MD, Pilar Cristancho, MD, and Andrew D. Peshek, MD “Vagus Nerve Stimulation (VNS) and Treatment of Depression: To the Brainstem and Beyond” Psychiatry (Edgmont). 2006 May; 3(5): 54–63.
- Yang Y, Yang LY, at al “Non-invasive vagus nerve stimulation reduces blood-brain barrier disruption in a rat model of ischemic stroke” Brain Stimul. 2018 Jul - Aug;11(4):689-698.
- Beatriz O. Amorim, Luciene Covolan, at al “Deep brain stimulation induces antiapoptotic and anti-inflammatory effects in epileptic rats” J Neuroinflammation. 2015; 12: 162.
- Kwiatek-Majkusiak J., Geremek M at al “Higher serum levels of pro-hepcidin in patients with Parkinson’s disease treated with deep brain stimulation” Neuroscience Letters. 19 June 2018.
- Kuti Baruch, Aleksandra Deczkowska, at al “PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease” Nature medicine. 18 January 2016.
- Hannah X Chu, Thiruma V Arumugam, “Role of CCR2 in inflammatory conditions of the central nervous system” J Cereb Blood Flow Metab. 2014 Sep; 34(9): 1425–1429.
Телеграм: t.me/ainewsline
Источник: medach.pro