Прионы, кальций, микробиота, пищевые гормоны и болезнь Альцгеймера
МЕНЮ
Главная страница
Поиск
Регистрация на сайте
Помощь проекту
Архив новостей
ТЕМЫ
Новости ИИ
Городские сумасшедшие
ИИ в медицине
ИИ проекты
Искусственные нейросети
Искусственный интеллект
Слежка за людьми
Угроза ИИ
ИИ теория
Компьютерные науки
Машинное обуч. (Ошибки)
Машинное обучение
Машинный перевод
Нейронные сети начинающим
Психология ИИ
Реализация ИИ
Реализация нейросетей
Создание беспилотных авто
Трезво про ИИ
Философия ИИ
Генетические алгоритмы
Капсульные нейросети
Основы нейронных сетей
Промпты. Генеративные запросы
Распознавание лиц
Распознавание образов
Распознавание речи
Творчество ИИ
Техническое зрение
Чат-боты
Авторизация
2019-09-21 17:00
Болезнь Альцгеймера (AD) является наиболее распространенной формой деменции у пожилых людей с клиническими проявлениями прогрессирующих когнитивных и функциональных нарушений. Сегодня только по официальным данным в мире насчитывается 47,5 миллионов человек с деменцией. Ежегодно регистрируется 7,7 млн. новых случаев заболевания. И ожидается, что число людей с деменцией увеличится до 75,6 млн. человек к 2030 году, а к 2050 — 135,5 млн. человек. На сегодня основным фактором повышения риска возникновения AD считается возраст.
Патология AD обычно характеризуется внеклеточными скоплениями ?-амилоидных пептидов (A?) в сенильных бляшках и внутриклеточными отложениями гиперфосфорилированного тау-белка, который образует нейрофибриллярные клубки. Генетически, аллели аполипопротеина E (APOE) (?2, ?3 и ?4) несут различные риски для развития AD. Лица с аллелем ?-4 имеют повышенный риск по сравнению с лицами с более распространенным аллелем ?3, в то время как аллель ?2 связан со сниженным риском [1]. Не так давно выяснилось, что патогенез AD может иметь долгую историю и отложение амилоида в мозге может предшествовать клиническим симптомам на 10–20 лет [2]. Имеются доказательства того, что AD связана с хроническим воспалением как в центральной нервной системе, так и на периферии [3-5]. Несмотря на длительное изучение, лечения AD пока не существует. Все попытки создания эффективной терапии пока потерпели неудачу. Поэтому учёные ищут новые подходы к этой болезни.
Кальций и дисфункция митохондрий
Так, в августе этого года в Nature Communications вышла статья John W. Elrod и соавт., “Нарушение митохондриального оттока кальция способствует прогрессированию заболевания в моделях болезни Альцгеймера”. В ней авторы на основе полученных результатов выдвинули гипотезу о первостепенном значении дисфункции митохондрий и перегрузки кальцием в развитии болезни Альцгеймера [6].
Дальше – цитата из релиза Temple University Health System к этой статье.
«Иногда, чем больше человек пытается решить, казалось бы, незначительную проблему, тем хуже становится. Оказывается, клетки действуют подобно, и попытка компенсировать то, что начинается как незначительный дефицит или дисфункция, может стать ужасной.
В случае болезни Альцгеймера исследователи показали, что ремоделирование транспорта митохондриального кальция — то, что, по-видимому, является попыткой клеток компенсировать пониженную выработку энергии и метаболическую дисфункцию – изначально может быть полезно, в конечном итоге становится неадаптивным, способствуя снижению митохондриальной функции и конгитивных способностей.
Новое исследование, опубликованное в журнале Nature Communications, является первым, которое связывает неадаптивные изменения в транспорте кальция в митохондриях — генерирующими энергию в клетках — с прогрессированием болезни Альцгеймера.
Вот как объясняет суть открытия руководитель исследования, John W. Elrod: «Отложение бета-амилоида и тау-патология считаются основными причинами болезни Альцгеймера и, как следствие, они являются основным направлением развития терапии. Однако крупные клинические испытания, нацеленные на эти пути, повсеместно провалились».
«Но до сих пор никто не исследовал влияние измененного транспорта кальция
в и из митохондрий на прогрессирование болезни Альцгеймера. Наше текущее исследование обеспечивает недостающую связь между этими двумя гипотезами патогенеза болезни Альцгеймера».
Транспорт кальция в митохондрии играет важную роль во многих клеточных функциях и требует эффективного участия множества белков. Одним из ключевых регуляторов этого процесса — белок, известный как NCLX, который ранее был обнаружен в лаборатории J. Elrod как участник регуляции транспорта кальция из клеток сердца. Экспрессия NCLX также важна для митохондриального транспорта кальция в нейронах.
В своем новом исследовании John W. Elrod и его коллеги изучили роль поглощения митохондриального кальция нейронами при болезни Альцгеймера. Для этого команда использовала мышиную модель болезни Альцгеймера, в которой животные обладали тремя генными мутациями, которые вызывают возрастную патологию, сравнимую с прогрессией болезни Альцгеймера у людей.
По мере старения мышей, несущих три мутации, исследователи наблюдали устойчивое снижение экспрессии NCLX. Это снижение сопровождалось снижением экспрессии белков, которые ограничивают накопление митохондриального кальция, что приводит к разрушительной перегрузке кальцием. Потеря NCLX была также связана с увеличением производства разрушающих клетки окислителей.
Чтобы лучше понять физиологическую значимость потери NCLX, учёные полностью подавили экспрессию NCLX в мозге мышей с болезнью Альцгеймера. Анализ мозговой ткани этих мышей показал, что снижение NCLX и последующая потеря оттока кальция из митохондрий ускоряют накопление бета-амилоида и тау-патологию.
«Наши результаты показывают, что дезадаптивное ремоделирование путей для компенсации нарушений регуляции кальция, которые, возможно, предназначены для поддержания выработки энергии в клетках, приводят к дисфункции нейронов и патологии болезни Альцгеймера. Более того, наши данные свидетельствуют о том, что накопление бета-амилоида и тау-патология на самом деле лежат ниже митохондриальной дисфункции в прогрессировании болезни Альцгеймера, что открывает новое терапевтическое направление». [7]
Кишечная микробиота и рацион
Кроме митохондрий и кальция, возможно диета и микробиота кишечника также существенно влияют на развитие AD. Начиная с 2014 года, исследователи показывали потенциальную роль патогенных микробов, в том числе кишечных, в развитии и прогрессировании AD. Как сегодня уже известно, кишечная микробиота – важная составляющая здоровья человека. Кишечные микробы выполняют ключевые функции для здоровья человека, включая извлечение энергии, биосинтез витаминов, защиту от чрезмерного роста патогенов и др. Микробная колонизация кишечника происходит во время родов, очень динамична в младенчестве и складывается во взрослую структуру примерно к 3 годам жизни.
Установлено, что изменения в составе сложной микробной экосистемы связаны с развитием различных желудочно-кишечных и метаболических заболеваний, включая воспалительные заболевания кишечника, ожирение, резистентность к инсулину и диабет [8]. В последнее время большое внимание уделяется влиянию кишечной микробиоты на функцию центральной нервной системы, которую часто называют осью кишечник — головной мозг. Изменения в микробиоме кишечника связаны с неврологическими состояниями, включая расстройства аутистического спектра, рассеянный склероз и Болезнь Паркинсона [9-11].
Исследования на животных моделях AD показали, что изменение микробиоты кишечника может влиять на скорость отложения амилоида [12,13]. У людей с AD учёные наблюдали уменьшение микробного разнообразия кишечного микробиома в отличие от группы контроля, состоящей из людей, соответствующих по возрасту и полу. Были выявлены различия между типами бактерий, включая уменьшение Firmicutes и Actinobacteria (в том числе и Bifidobacteriaceae) и увеличение Bacteroidetes в микробиоме людей с AD [14].
Вместе с другими факторами, рацион питания может играть свою роль в патологии AD. Повышенный риск развития деменции связывают с рационом питания с высоким содержанием насыщенных жиров и простых углеводов, то, что называется «западной» диетой. И напротив, диеты с высоким содержанием моно- и полиненасыщенных жиров, овощей, фруктов и нежирных белков связаны с пониженным риском AD [15 — 17].
В 2013 году ЮНЕСКО включила средиземноморскую диету в «Репрезентативный список нематериального культурного наследия человечества» [18]. Диета в средиземноморском стиле представляет собой сбалансированное питание, характеризующееся высоким потреблением клетчатки, оливкового масла, фруктов, орехов, овощей и злаков; умеренным потреблением рыбы и птицы; низким потреблением молочных продуктов, красного мяса, мясных полуфабрикатов и сладостей. А также потреблением вина в умеренных количествах [19]. Было обнаружено, что средиземноморская диета связана с уменьшением частоты некоторых хронических заболеваний, таких как ожирение, диабет 2 типа, рак, сердечно-сосудистые и нейродегенеративные заболевания, включая болезнь Альцгеймера [17, 20, 21].
Хотя лежащие в основе этого механизмы остаются не вполне ясными, последние данные свидетельствуют о том, что модуляция кишечного микробиома и микробных метаболитов является одним из возможных факторов, опосредующих воздействие средиземноморской диеты на здоровье [20, 22].
У людей с умеренными конгитивными нарушениями учёные обнаружили, что определённые бактерии связаны с маркерами болезни Альцгеймера в спинномозговой жидкости. Модифицированная средиземноморская кетогенная диета (MMKD) меняла кишечный микробиом и снижала уровни одного из главных маркеров AD, A?42. MMKD была урезана по углеводам, при этом главную часть рациона составляли жиры и белки, полученные из оливкового масло и рыбы. Несколько механизмов действия кетогенной диеты, включая снижение гипервозбудимости нейронов, усиление митохондриального метаболизма, снижение окислительного стресса и ингибирование mTOR, могут оказывать влияние на патологические процессы при AD [23].
После 6 недель диеты MMKD у участников с умеренными когнитивными нарушениями, которые часто предшествуют AD, были отмечены снижение содержания лактата и повышение содержания бутирата, а также позитивные изменения в кишечном микробиоме, и снижение уровня в спинномозговой жидкости A?42 [24].
В подтверждении о важной роли кишечной микробиоты в патогенезе AD, в этом году вышло сразу три исследования по трансплантации фекальной микробиоты. Нормальную микробиоту кишечника от здоровых мышей пересаживали трансгенным мышам с моделью AD. Во всех случаях происходило заметное улучшение состояния животных с AD: «Наши результаты показали, что применение FMT (трансплантации фекальной микробиоты) может улучшить когнитивный дефицит и уменьшить отложение в мозге ?-амилоида (A?) у трансгенных (Tg) мышей APPswe / PS1dE9. Эти улучшения сопровождались снижением фосфорилирования тау-белка и уровней A?40 и A?42. Мы наблюдали увеличение синаптической пластичности у мышей Tg, при этом экспрессия белка PSD-95 и экспрессия синапсина I увеличивались после FMT. Мы также наблюдали снижение уровней COX-2 и CD11b у мышей Tg после FMT. Также мы обнаружили, что применение FMT вызвало изменения кишечной микробиоты и SCFAs (короткоцепочечных жирных кислот). Таким образом, FMT может быть потенциальной терапевтической стратегией для AD» [42-44].
Прионная природа нейропатологий
Также в этом году пришла ещё одна важная новость по болезни Альцгеймера. Было опубликовано первое проспективное исследование, показавшее повышение риска возникновения AD после переливания крови. В исследуемую группу вошли 63 813 пациентов, перенесших переливание крови, и столько же человек контрольной группы. Период наблюдения составил 10 лет. Полученные результаты показали, что у людей, перенесших переливание крови, риск развития деменции в 1,73 раза выше и в 1,37 раза выше риск возникновения болезни Альцгеймера, чем у тех, кто этого не сделал. Пациенты, которые получали переливание отмытых эритроцитов, имели в 2,37 раза более высокий риск развития деменции по сравнению с теми, кто этого не делал [45].
Хотя авторы осторожны в своих выводах, одной из возможных причин этой важного открытия может выступать прионная природа болезни Альцгеймера. И об этом стоит рассказать поподробнее.
Одна из первых прорывных работ по этой теме вышла ещё в 2015 году в журнале Nature. Sebastian Brandner с коллегами обследовали посмертные образцы мозга людей, умерших от болезни Крейтцфельдта-Якоба, или «коровьего бешенства». Эта болезнь имеет прионную природу. Люди, мозг которых исследовали учёные, заразились в результате инъекций инфицированного прионами гормона роста. Они имели средний возраст, 36-51 год, и кроме прионов ткань их мозга содержала скопления бета-амилоида. Все эти люди не имели генетической предрасположенности к болезни Альцгеймера. Кроме того, учёные обследовали 116 пациентов с другими прионными заболеваниями такого же и старше на 10 лет возраста. Ни у одного из обследованных не было выявлено скоплений бета-амилоида в мозге. Что позволило учёным предположить, что бета-амилоид появился в мозге людей с болезнью Крейтцфельдта-Якоба по той же причине, что и прионы – в результате заражения инфицированными инъекциями гормонов роста [46].
В 2018 году вышло ещё две работы этой команды учёных. Nature опубликовал их статью, в которой они сообщали результаты исследования образцов гормонов роста, полученных из гипофиза человека, которые вызывали развитие прионной болезни. «Учитывая важность нашей гипотезы для общественного здравоохранения, мы приступили к идентификации и биохимическому анализу архивных флаконов c-hGH (гормона роста). Здесь мы показываем, что определенные партии c-hGH, которые применялись пациентами с патологией Крейтцфельда-Якоба и скоплениями A?, имеют существенные уровни A?40, A? 42 и тау-белков. И этот материал может вызывать образование бляшек A? и церебральную амилоидную ангиопатию у мышей, экспрессирующих человеческий белок-предшественник амилоида. Эти результаты подтверждают наличие A? в архивированных флаконах c-hGH и согласуются с предполагаемой ятрогенной ( т.е. вызванной посредством медицинских процедур) передачей патологии A? человеку» – резюмируют авторы исследования [47].
Во второй работе этого же года S. Brandner и соавт. предоставили данные, показывающие возможность заражения бета-амилоидами во время нейрохирургических операций. «Здесь мы представляем четырех пациентов, которые перенесли нейрохирургические процедуры в детском или подростковом возрасте и получили примерно три десятилетия спустя внутримозговое кровоизлияние, вызванное тяжелой церебральной амилоидной ангиопатией (CAA). Ни у одного из этих пациентов не было патогенных мутаций, связанных с ранним развитием патологии A?. Кроме того, мы выявили в литературе четырех пациентов с нейрохирургическим вмешательством в анамнезе и последующим развитием CAA» [48].
За год до этого вышло ставшее настоящей сенсацией громкое исследование китайских учёных. Здоровым и трансгенным мышам с AD объединили кровоток, в результате чего у диких нормальных животных в мозге появились скопления бета-амилоида. Фактически произошло заражение амилоидом через кровь от больного животного к здоровому. «В этом исследовании, используя модель парабиоза между трансгенными мышами AP APsswe / PS1dE9 и их дикими собратьями, мы обнаружили, что человеческий A?, происходящий от трансгенных мышей модели AD, попал в кровообращение и накопился в мозге мышей дикого типа, и сформировалась церебральная амилоидная ангиопатия и бляшки A? после 12-месячного периода парабиоза. Патологии типа AD, связанные с накоплением A?, включая гиперфосфорилирование тау, нейродегенерацию, воспаление и микрогеморрагии были обнаружены в мозге парабиотических мышей дикого типа. Насколько нам известно, наше исследование является первым, которое выявило, что происходящий из крови A? может проникать в мозг, формировать связанные с A? патологии и вызывать функциональный дефицит нейронов» [49].
Весомости данной гипотезе, о возможности заражения и дальнейшего распространения бета-амилоида по прионному принципу придаёт то, что её поддерживает первооткрыватель прионов, Нобелевский лауреат Стенли Прузинер. В 2019 году в Science Translational Medicine вышла работа С. Прузинера и его коллег, в которой они предоставили новые доказательства этой гипотезы [50].
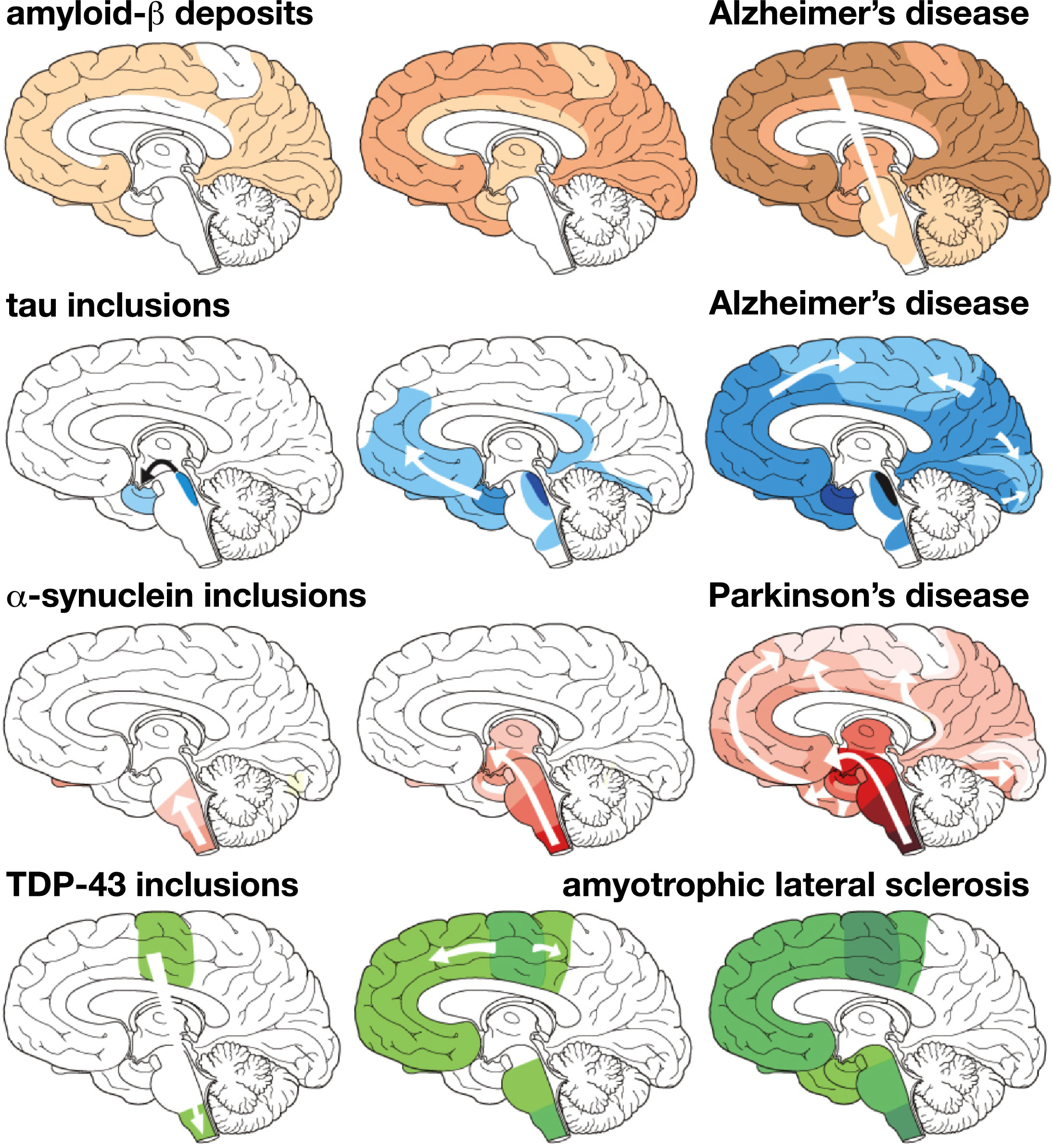
Что обращает на себя внимание: вторая и третья по распространённости нейропатологии после AD, болезнь Паркинсона и боковой амиотрофический склероз, также возможно распространяются по организму прионоподобным образом. И об этом имеется целый ряд исследований [51, 52]. Так, скопления альфа-синуклеина, главного участника болезни Паркинсона, как было описано в недавней работе 2019 года, вначале появляются в кишечнике и затем продвигаются по другим тканям подобно прионам. Причём, эти скопления в желудочно-кишечном тракте могут обнаруживаться, как говорят учёные, за 20 лет до появления клинических симптомов болезни Паркинсона. “В этом новом исследовании мы выяснили, как именно болезнь может распространяться из кишечника людей. Мы, вероятно, не сможем разработать эффективные методы лечения, которые остановят болезнь, не зная, где она начинается и как она распространяется — так что это важный шаг в нашем исследовании патологии. Болезнь Паркинсона — это сложное заболевание, которое мы все еще пытаемся понять. Однако, благодаря этому исследованию и аналогичному исследованию в США, которое недавно привело к тому же результату с использованием мышей, возникает предположение, что заболевание начинается в кишечнике пациентов” — говорит один из авторов работы, P. Borghammer [53].
Интересные данные о сходных «родственных» механизмах патогенеза болезней Альцгеймера и Паркинсона были получены Z. Zhang и соавт. Они показали, что один и тот же фермент, аспарагин-эндопептидаза (AEP), расщепляет как тау-белок при AD, так и альфа-синуклеин при БП, что приводит к их агрегации и нейротоксичности [54].
Вполне очевидно, что прионоподобный характер распространения трёх основных нейродегенеративных патологий вряд ли может быть случайностью. Хотя возможность заражения извне показана сегодня только для болезни Альцгеймера. В 2019 году американские учёные, J. Weickenmeier и соавт., смогли подвести теоретическую базу под прионную гипотезу для этих трёх нейропатологий. Они создали для них физическую модель, которая объясняет прионоподобные особенности нейродегенерации при болезни Альцгеймера, Паркинсона и боковом амиотрофическом склерозе: «Наши результаты показывают, что неправильно свернутые белки при различных нейродегенеративных заболеваниях растут и распространяются в соответствии с универсальным законом, который следует основным физическим принципам нелинейной реакции и анизотропной диффузии. Наши результаты подтверждают понятие общего основополагающего принципа патогенеза широкого спектра нейродегенеративных расстройств — прионной парадигмы». Любители сложных формул и уравнений смогут получить большое удовольствие от прочтения данной статьи [55].
Ну и в дополнении к прионам, расскажем ещё об одной обнаруженной интересной особенности мозга людей с AD. Как показали британские учёные, Shelley J. Allen и соавт., мозговая ткань пациентов с AD содержит аномально большое количество микробов с перекосом в сторону P. acnes. "P. acnes является комменсальным грамположительным компонентом микрофлоры кожи и рта человека, предпочитая анаэробные условия роста, и становится все более очевидным, что он является значительным условно-патогенным микроорганизмом. Чаще всего это связано с послеоперационными поражениями и имплантированными протезами, а также с хроническими заболеваниями, такими как воспаление поясничного отдела, эндокардит, саркоидоз и внутричерепные поражения. Недавно было показано наличие протеобактерий и актинобактерий (содержащих Propionibacteriaceae ) в мозге, пораженном как нормальным, так и рассеянным склерозом; таким образом, нормальный мозг имеет микробиом, состоящий в основном из протеобактерий и актинобактерий. Наши данные свидетельствуют о том, что Actinobacteria ( P. acnes) увеличивается в головном мозге AD в ущерб протеобактерий. Способность P. acnes неспецифически стимулировать врожденную иммунную систему хорошо документирована" — пишут авторы работы [56].
“Пищевые” гормоны в патогенезе AD
Также в недавней работе учёные нашли, что гормон голода грелин возможно участвует в развитии болезни Альцгеймера. Рецепторы грелина не так давно были обнаружены в гиппокампе. Гиппокамп — это область мозга, важная для обучения, памяти и эмоций. При AD, это одна из первых областей, где происходит гибель и повреждение клеток из-за образования скоплений бета-амилоида. В здоровом гиппокампе грелин связывается со своим рецептором, GHSR1?. Затем это опосредует активацию дофаминового рецептора D1 (DRD1). Модуляция DRD1 с помощью GHSR1? является критической для функции синапсов гиппокампа и синаптической реорганизации посредством сигнального пути G?q-Ca 2+, который является центральным для формирования памяти.
Роль GHSR1? в синаптической функции гиппокампа позволила учёным предположить, что дисфункция рецептора может способствовать синаптическому дефициту в гиппокампе, наблюдаемому при AD.
Исследователи из Университета Далласа изучили этот вопрос, используя образцы мозга людей, страдавших AD и модель животных с AD. Гипотеза учёных состояла в том, что диссоциация (то есть разделение) между рецепторами грелина и дофамина может быть тем самым фактором, который влияет на познавательную способность у пациентов с болезнью Альцгеймера.
Ученым удалось обнаружить, что ?-амилоид подавляет активацию GHSR1?. Что в свою очередь, нарушает GHSR1?-опосредованную активацию DRD1 в гиппокампе людей с AD. На животной модели исследователи вводили мышам с AD два вещества, активирующие рецепторы грелина и дофамина в гиппокампе, MK0677 и SKF8129. Эта комбинация предотвращала ингибирование рецептора GHSRr1? ?-амилоидом, смягчая синаптическое повреждение гиппокампа и улучшая обучение и память грызунов [25].
Что характерно, грелин – не первый «пищевой» гормон, который может быть связан с когнитивными нарушениями и альцгеймером. До этого обнаружили влияние инсулина с резистентностью к нему, даже назвав Альцгеймер диабетом 3 типа.
Как показывают имеющиеся данные, AD может быть медленно прогрессирующим метаболическим заболеванием головного мозга. А многочисленные исследования демонстрируют сложную связь между метаболическим синдромом (MetS) и AD. Люди с сахарным диабетом и ожирением имеют более высокий риск развития AD. В то же время у пациентов с AD часто развивается гипергликемия и инсулинорезистентность (IR). IR как нарушение передачи сигналов инсулина является общей характерной особенностью как MetS, так и AD. И, по мнению учёных, представляет собой ключевое связующее звено между двумя заболеваниями. Передача сигналов инсулина регулирует уровни A? и тау, а A? оказывает негативное влияние на передачу сигналов инсулина. Следовательно, дисфункция передачи сигналов инсулина может усиливать патологию A? и тау, а повышенная продукция A? может дополнительно усугублять IR. Накопленные данные также свидетельствуют о том, что AD тесно связана с дисфункцией как передачи сигналов инсулина, так и метаболизма глюкозы в мозге, что побуждает некоторых исследователей относить AD к диабету 3 типа или к инсулинрезистентному состоянию мозга [26, 27].
Инсулин, секретируется бета-клетками поджелудочной железы и проникает в центральную нервную систему, пересекая гематоэнцефалический барьер регулируемым и насыщаемым образом. Синтез инсулина в самом мозге является пока предметом дискуссий [28]. Рецепторы инсулина (InsRs) широко экспрессируются в мозге, в том числе в обонятельной луковице, коре головного мозга, гиппокампе, гипоталамусе и миндалине [29]. InsRs более сконцентрированы в нейронах по сравнению с глиальными клетками [30].
Передача сигналов инсулина в мозг играет важную роль в регуляции потребления пищи, массы тела, репродукции, обучения и памяти. Интраназальное введение инсулина улучшает рабочую память как в исследованиях на людях, так и на животных [31]. Кроме того, уровни мРНК и белка InsR увеличиваются в области СА1 гиппокампа в процессах формирования кратковременной памяти [32]. Что позволяет предположить, что чувствительность нейронов к инсулину может быть повышена во время обучения.
Нарушение передачи сигналов инсулина, делает нейроны более уязвимыми для метаболического стресса, ускоряя нейронную дисфункцию. Дефектная передача сигналов инсулина связана со снижением когнитивных способностей и развитием деменции, включая AD [33]. Ухудшение когнитивных способностей при диабете и AD связано со снижением экспрессии InsR и уровня инсулина в спинномозговой жидкости (CSF) [34, 35]. Снижение передачи сигналов инсулина, включая измененную активность киназы и экспрессию IRS, при AD ухудшается с прогрессированием заболевания [36,37]. И повышение фосфорилирования IRS-1, ключевого фактора IR, происходит в мозге при AD [38]. Интересно, что области мозга с самой высокой плотностью InsR, такие как гиппокамп и височная доля, также являются основными мишенями нейродегенерации при AD [39, 40]. Следовательно, нарушение передачи сигналов инсулина, вызванное IR, может оказывать большое влияние на снижение когнитивных функций и развитие AD.
Несколько методов лечения диабета, которые усиливают передачу сигналов инсулина, тестируются на терапевтический потенциал при AD и деменции. И хотя результаты клинических испытаний TZD оказались неутешительными, интраназальный инсулин и аналоги GLP-1 все еще активно используются в качестве потенциального лечения AD и показали некоторые многообещающие результаты. Интраназальный инсулин, однако, эффективен только на ранних стадиях AD. Кроме того, эксенатид и лираглутид все еще находятся на ранних стадиях терапевтического развития, и в настоящее время проводятся крупные клинические испытания [41].
Но и это не всё. Выясняется, что кроме грелина и инсулина, ещё два пищевых гормона, из числа адипокинов (гормонов жировой ткани), лептин и адипонектин могут участвовать в Альцгеймере.
Есть свежие исследования этого года:
Ожирение как фактор риска болезни Альцгеймера: влияние лептина и глутамата Роль лептина и адипонектина в когнитивном снижении, связанном с ожирением, и болезни Альцгеймера. Более ранние работы, 2016 и 2018 года:
Дисфункция лептина и болезнь Альцгеймера: данные клеточных исследований, исследований на животных и людях. Лептиновая регуляция функции гиппокампа и ее роль в болезни Альцгеймера.
Появляется все больше свидетельств того, что лептин обладает когнитивно-стимулирующими свойствами, так как он участвует в сигнальных путях, лежащих в основе гиппокампального обучения и памяти. Тем не менее, значительное снижение способности лептина регулировать синаптическую функцию гиппокампа возникает с возрастом, и дисфункции в лептиновой системе связаны с повышенным риском развития болезни Альцгеймера.
www.ncbi.nlm.nih.gov/pubmed/28987937 Ещё один гормон, связанный с жировой тканью и воспалением, резистин, играет одну из важных ролей в воспалении гипоталамуса и нарушении гормональной регуляции:
Молекулярные механизмы, лежащие в основе индуцированного ожирением воспаления гипоталамуса и инсулинорезистентности: ключевая роль пути резистина / TLR4
www.ncbi.nlm.nih.gov/pmc/articles/PMC6418006 В итоге что получается? Целый ряд “пищевых гормонов” (грелин, инсулин, резистин, лептин, адипонектин), регуляция которых нарушается в процессе старения, могут так или иначе участвовать в нейродегенеративных процессах.
Поэтому вообще не случайно, что антидиабетические препараты, метформин и другие, проходят доклинические исследования на предмет противодействия возрастной нейродегенерации.
Противодиабетические препараты при болезни Альцгеймера: механизмы действия и перспективы на будущее Ну и в самом конце коротко скажем, как нарушение режима и качества сна может быть связано с развитием AD. В 2018 году в PNAS вышла работа N. Volkow и соавт., в которой они показали, что даже одна бессонная ночь может увеличить накопление бета-амилоида в мозге [57].
И в 2019 году учёные из Вашингтонского университета опубликовали работу, показавшую, что снижение глубокого сна связано с ранними признаками болезни Альцгеймера [58].
Подведём итог. Самые распространённые возрастные нейродегенеративные патологии показывают чрезвычайно сложную природу своего патогенеза. Понять которую и сложить в единую картину мы пока не в силах. Эффективных лекарств против них пока нет и даже не понятно, на какой основе их создавать. Поэтому, то, что человек может сейчас сделать — это вести здоровый образ жизни и поддерживать по мере возможности работы и исследования, направленные против старения в целом и возрастной нейродегенерации в частности.
Обзор подготовили: М. Батин, А. Ржешевский.
1.Liu, C.C., Liu, C.C., Kanekiyo, T., Xu, H., and Bu, G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013; 9: 106–118
2. Penke, B., Bogar, F., and Fulop, L. Beta-amyloid and the Pathomechanisms of Alzheimer's disease: a comprehensive view. Molecules (Basel, Switzerland). 2017; 22: 10
3. Le Page, A., Dupuis, G., Frost, E.H., Larbi, A., Pawelec, G., Witkowski, J.M. et al. Role of the peripheral innate immune system in the development of Alzheimer's disease. Exp Gerontol. 2018; 107: 59–66
4. Bronzuoli, M.R., Iacomino, A., Steardo, L., and Scuderi, C. Targeting neuroinflammation in Alzheimer's disease. Journal of inflammation research. 2016; 9: 199–208
5. Kagan, B.L., Jang, H., Capone, R., Teran Arce, F., Ramachandran, S., Lal, R. et al. Antimicrobial properties of amyloid peptides. Mol Pharm. 2011; 9: 708–717
6. Jadiya P, Kolmetzky DW, Tomar D, Di Meco A, Lombardi AA, Lambert JP, Luongo TS, Ludtmann MH, Pratic? D, Elrod JW. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer's disease. Nat Commun. 2019 Aug 29;10(1):3885.
7. Temple Scientists Identify Promising New Target to Combat Alzheimer’s Disease. www.templehealth.org/about/news/temple-scientists-identify-promising-new-target-to-combat-alzheimers-disease 8. Clemente, J. C., Ursell, L. K., Parfrey, L. W. & Knight, R. The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 (2012). 9. Fung, T. C., Olson, C. A. & Hsiao, E. Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 20, 145–155 (2017). 10.Scheperjans, F. et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 30, 350–358 (2015).
11.Keshavarzian, A. et al. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 30, 1351–1360 (2015).
12. Cattaneo, A. et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49, 60–68 (2017).
13. Minter, M. R. et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 6, 1–12 (2016).
14. Harach, T. et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 7, 41802 (2017).
15. Morris, M.C. and Tangney, C.C. Dietary fat composition and dementia risk. Neurobiol Aging. 2014; 35: S59–S64
16. Gu, Y. and Scarmeas, N. Dietary patterns in Alzheimer's disease and cognitive aging. Curr Alzheimer Res. 2011; 8: 510–519.
17. Chianese, R., Coccurello, R., Viggiano, A., Scafuro, M., Fiore, M., Coppola, G. et al. Impact of dietary fats on brain functions. Curr Neuropharmacol. 2018; 16: 1059–1085
18. Bach-Faig, A., Berry, E.M., Lairon, D., Reguant, J., Trichopoulou, A., Dernini, S. et al. Mediterranean diet pyramid today. Science and cultural updates. Public Health Nutr. 2011; 14: 2274–2284
19. Willett, W.C., Sacks, F., Trichopoulou, A., Drescher, G., Ferro-Luzzi, A., Helsing, E. et al. Mediterranean diet pyramid: a cultural model for healthy eating. (Suppl)Am J Clin Nutr. 1995; 61: 1402S–1406S
20. Tosti, V., Bertozzi, B., and Fontana, L. Health benefits of the Mediterranean diet: metabolic and molecular mechanisms. J Gerontol A Biol Sci Med Sci. 2018; 73: 318–326
21. Sofi, F., Abbate, R., Gensini, G.F., and Casini, A. Accruing evidence on benefits of adherence to the Mediterranean diet on health: an updated systematic review and meta-analysis. Am J Clin Nutr. 2010; 92: 1189–1196
22. David, L.A., Maurice, C.F., Carmody, R.N., Gootenberg, D.B., Button, J.E., Wolfe, B.E. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014; 505: 559–563
23. Ma, D., Wang, A.C., Parikh, I., Green, S.J., Hoffman, J.D., Chlipala, G. et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci Rep. 2018; 8: 6670
24. Nagpal R1, Neth BJ2, Wang S1, Craft S3, Yadav H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer's disease markers in subjects with mild cognitive impairment. EBioMedicine. 2019 Aug 30. pii: S2352-3964(19)30554-7.
25. Tian J et al. Disrupted hippocampal growth hormone secretagogue receptor 1? interaction with dopamine receptor D1 plays a role in Alzheimer's disease. Sci Transl Med. 2019 Aug 14;11(505). pii: eaav6278.
26. Frisardi V, Solfrizzi V, Capurso C, Imbimbo BP, Vendemiale G, Seripa D et al. Is insulin resistant brain state a central feature of the metabolic-cognitive syndrome? J Alzheimers Dis 2010; 21: 57–63.
27. Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. J Alzheimers Dis 2006; 9: 13–33.
28. Baskin DG, Figlewicz DP, Woods SC, Porte D Jr ., Dorsa DM. Insulin in the brain. Annu Rev Physiol 1987; 49: 335–347.
29. Unger JW, Livingston JN, Moss AM. Insulin receptors in the central nervous system: localization, signalling mechanisms and functional aspects. Prog Neurobiol 1991; 36: 343–362.
30.van der Heide LP, Ramakers GM, Smidt MP. Insulin signaling in the central nervous system: learning to survive. Prog Neurobiol 2006; 79: 205–221.
31. Benedict C, Frey WH 2nd, Schioth HB, Schultes B, Born J, Hallschmid M. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp Gerontol 2011; 46: 112–115.
32. Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ et al. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem 1999; 274: 34893–34902.
33.de la Monte SM. Insulin resistance and Alzheimer's disease. BMB Rep 2009; 42: 475–481.
34. Duarte AI, Moreira PI, Oliveira CR. Insulin in central nervous system: more than just a peripheral hormone. Journal of aging research 2012; 2012: 384017.
35. Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 2010; 31: 224–243.
36. Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J Cell Mol Med 2011; 15: 1807–1821.
36. Li L, Holscher C. Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev 2007; 56: 384–402.
37. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 2012; 122: 1316–1338.
38. de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer's disease. J Alzheimers Dis 2005; 7: 45–61.
39. Freude S, Schilbach K, Schubert M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer's disease: from model organisms to human disease. Curr Alzheimer Res 2009; 6: 213–223.
40. Gammeltoft S, Fehlmann M, Van Obberghen E. Insulin receptors in the mammalian central nervous system: binding characteristics and subunit structure. Biochimie 1985; 67: 1147–1153.
41. Bhumsoo Kim, Eva L Feldman. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome Exp Mol Med. 2015 Mar; 47(3): e149. Published online 2015 Mar 13.
42. Jing Sun et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl Psychiatry. 2019; 9: 189.
43. Shalini Elangovan, Thomas J Borody, Damian Holsinger. Fecal Microbiota Transplantation Decreases Amyloid Load and Improves Cognition in Alzheimer’s. BioRxiv Jul 1, 2019.
44. Kim M, Kim Y, Choi H, et al Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut. Published Online First: 30 August 2019. doi: 10.1136/gutjnl-2018-317431.
45. Shih-Yi Lin et al. Association of Transfusion With Risks of Dementia or Alzheimer’s Disease: A Population-Based Cohort Study Front Psychiatry. 2019; 10: 571.
46. Jaunmuktane Z et al. Evidence for human transmission of amyloid-? pathology and cerebral amyloid angiopathy. Nature. 2015 Sep 10;525(7568):247-50.
47. Purro SA et al. Transmission of amyloid-? protein pathology from cadaveric pituitary growth hormone. Nature. 2018 Dec;564(7736):415-419. doi: 10.1038/s41586-018-0790-y. Epub 2018 Dec 13.
48. Jaunmuktane Z et al. Evidence of amyloid-? cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol. 2018 May;135(5):671-679.
49. Bu XL et al. Blood-derived amyloid-? protein induces Alzheimer's disease pathologies. Mol Psychiatry. 2018 Sep;23(9):1948-1956.
50. Atsushi Aoyagi, Carlo Condello, Jan St?hr, Weizhou Yue, Brianna M. Rivera, Joanne C. Lee, Amanda L. Woerman, Glenda Halliday, Sjoerd Van Duinen, Martin Ingelsson, Lars Lannfelt, Caroline Graff, Thomas D. Bird, C. Dirk Keene, William W. Seeley, William F. Degrado, and Stanley B. Prusiner. A? and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Science Translational Medicine, 2019.
51. Ayers JI, Cashman NR. Prion-like mechanisms in amyotrophic lateral sclerosis.
Handb Clin Neurol. 2018;153:337-354.
52. Kujawska M, Jodynis-Liebert J.. What is the Evidence That Parkinson's Disease is a Prion Disorder, Which Originates in the Gut? Int J Mol Sci. 2018 Nov 12;19(11). pii: E3573
53.Van Den Berge N et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019 Jun 26.
54. Zhentao Zhang, Seong Su Kang, Xia Liu, Eun Hee Ahn, Zhaohui Zhang, Li He, P Michael Iuvone, Duc M Duong, Nicholas T Seyfried, Matthew J Benskey, Fredric P Manfredsson, Lingjing Jin, Yi E Sun, Jian-Zhi Wang, Keqiang Ye. Asparagine endopeptidase cleaves ?-synuclein and mediates pathologic activities in Parkinson's disease. Nature Structural & Molecular Biology, 2017; DOI: 10.1038/nsmb.3433
55. Johannes Weickenmeier, Mathias Juckerb, Alain Gorielyc, Ellen Kuh. A physics-based model explains the prion-like features of neurodegeneration in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis Author links open overlay panel. Journal of the Mechanics and Physics of Solids Volume 124, March 2019, Pages 264-281.
56. David C. Emery, Deborah K. Shoemark, Tom E. Batstone, Christy M. Waterfall, Jane A. Coghill, Tanya L. Cerajewska, Maria Davies, Nicola X. West, Shelley J. Allen. 16S rRNA Next Generation Sequencing Analysis Shows Bacteria in Alzheimer’s Post-Mortem Brain. Frontiers in Aging Neuroscience, 2017; 9 DOI: 10.3389/fnagi.2017.00195
57. Shokri-Kojori E. et al. ?-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A. 2018 Apr 24;115(17):4483-4488.
58. Lucey BP, McCullough A, Landsness EC, Toedebusch CD, McLeland JS, Zaza AM, Fagan AM, McCue L, Xiong C, Morris JC, Benzinger TLS, Holtzman DM. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Science Translational Medicine, Jan. 9, 2019
Телеграм: t.me/ainewsline
Источник: habr.com