Калиевые каналы внутреннего выпрямления: часть 1
МЕНЮ
Главная страница
Поиск
Регистрация на сайте
Помощь проекту
Архив новостей
ТЕМЫ
Новости ИИ
Городские сумасшедшие
ИИ в медицине
ИИ проекты
Искусственные нейросети
Искусственный интеллект
Слежка за людьми
Угроза ИИ
ИИ теория
Компьютерные науки
Машинное обуч. (Ошибки)
Машинное обучение
Машинный перевод
Нейронные сети начинающим
Психология ИИ
Реализация ИИ
Реализация нейросетей
Создание беспилотных авто
Трезво про ИИ
Философия ИИ
Генетические алгоритмы
Капсульные нейросети
Основы нейронных сетей
Промпты. Генеративные запросы
Распознавание лиц
Распознавание образов
Распознавание речи
Творчество ИИ
Техническое зрение
Чат-боты
Авторизация
2019-08-07 20:16

В 1949 году Бернард Катц зарегистрировал в скелетных мышцах калиевый ток [1], который резко отличался от известного на тот момент калиевого тока в гигантском аксоне кальмара. Оказалось, что его амплитуда выше при значениях мембранного потенциала, отрицательных по отношению к равновесному потенциалу для калия (EK), чем при положительных (основы электрофизиологии кратко изложены в статье «Мембранный потенциал»).
Предпочтительное проведение тока только в одном направлении по аналогии с термином в электротехнике называют выпрямлением, а так как эти каналы пропускают преимущественно входящий ионный ток, их назвали калиевыми каналами внутреннего выпрямления, или Kir (inward rectifying K+-channels). Поскольку при физиологических концентрациях калия внутри и вне клетки уравнение Гольдмана — Ходжкина — Катца для тока предсказывает слабое внешнее выпрямление (рис. 1), эти каналы поначалу были описаны как каналы «аномального» выпрямления.
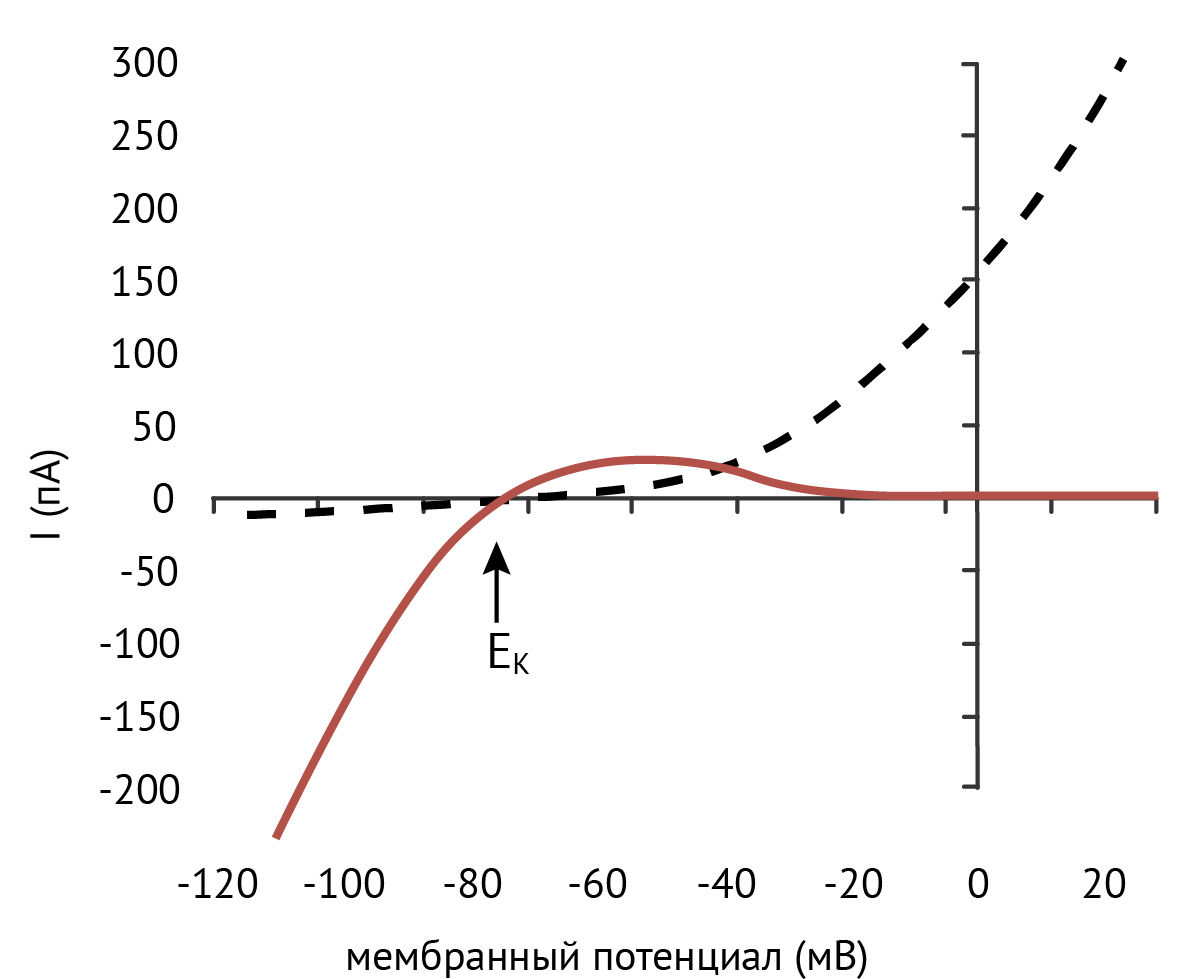
Рисунок 1
Зависимость силы тока от потенциала на мембране для каналов внутреннего (красная сплошная линия) и внешнего выпрямления (черная пунктирная линия). EK — равновесный потенциал для K+.
В физиологических условиях каналы Kir пропускают высокоамплитудный ток, когда мембрана гиперполяризована, и малоактивны при ее деполяризации. Эти каналы можно сравнить с диодами, проводящими ток в одном направлении. При высоком уровне экспрессии калиевых каналов внутреннего выпрямления потенциал покоя клеток будет близким к EK и в них не будет наблюдаться спонтанная электрическая активность.
Таким образом, электрофизиологические характеристики каналов Kir определяют их роль в поддержании потенциала покоя и регуляции возбудимости клеток. Мутации этих каналов могут приводить к патологии многих органов: аритмиям, заболеваниям сетчатки, периодическому параличу и сахарному диабету.
Структура каналов семейства Kir
Первичная структура калиевых каналов внутреннего выпрямления стала известна в 1993 году, когда были клонированы первые три представителя семейства Kir: классический IRK1/Kir2.1 [2], АТФ-зависимый ROMK1/Kir1.1 [3] и активируемый G-белками GIRK1/Kir3.1 [4].
Как можно установить соответствие между ионным током и геном, кодирующим ионный канал? Рассмотрим, как в своей работе K. Хо с коллегами клонировали первый канал этого семейства [3]. Сначала исследователи выделили полиаденилированную РНК из внутренней полоски мозгового вещества почки крысы и инъецировали ее в ооциты лягушки Xenopus laevis. В ооцитах, экспрессирующих эту РНК, методом фиксации потенциала с помощью двух электродов (описание метода см. в статье «Мембранный потенциал») можно зарегистрировать калиевый ток внутреннего выпрямления, отсутствующий в контрольных ооцитах, в которые ввели воду. Далее исследователи фракционировали использованную ранее тотальную РНК и инъецировали в ооциты получившиеся отдельные фракции. Таким образом они определили, что интересующий их транскрипт оказался в диапазоне 2–3 килобаз, и изготовили из этой фракции библиотеку комплементарной ДНК (кДНК). На основе клонов из библиотеки методом транскрипции in vitro была получена кРНК, которую затем экспрессировали в ооцитах лягушки. В итоге ученые обнаружили кРНК и, соответственно, кДНК длиной 2,1 килобаз, которая при экспрессии в ооцитах давала ток внутреннего выпрямления, блокируемый ионами Ba2+. Этот транскрипт назвали ROMK1 (renal outer medulla K channel 1). Затем кДНК секвенировали и таким образом определили первичную структуру канала.
Функциональный калиевый канал внутреннего выпрямления представляет собой гомо- или гетеротетрамер из Kir субъединиц. Базовая архитектура всех субъединиц Kir одинакова: NH2- и COOH-концы белка находятся в цитозоле, а трансмембранная часть состоит из двух трансмембранных доменов (TM1 и TM2) и порообразующего домена H5 между ними (см. рис. 2). Домен H5 служит селективным фильтром и содержит последовательность T-X-G-Y(F)-G, общую для всех семейств калиевых каналов. В отличие от потенциалзависимых калиевых, натриевых и кальциевых каналов (Kv, Nav и Cav), у Kir отсутствует чувствительный к мембранному потенциалу домен S4 и поэтому они открыты при любых значениях мембранного потенциала [5], если соблюдены другие условия открытия канала, например, низкий уровень АТФ для KATP каналов.
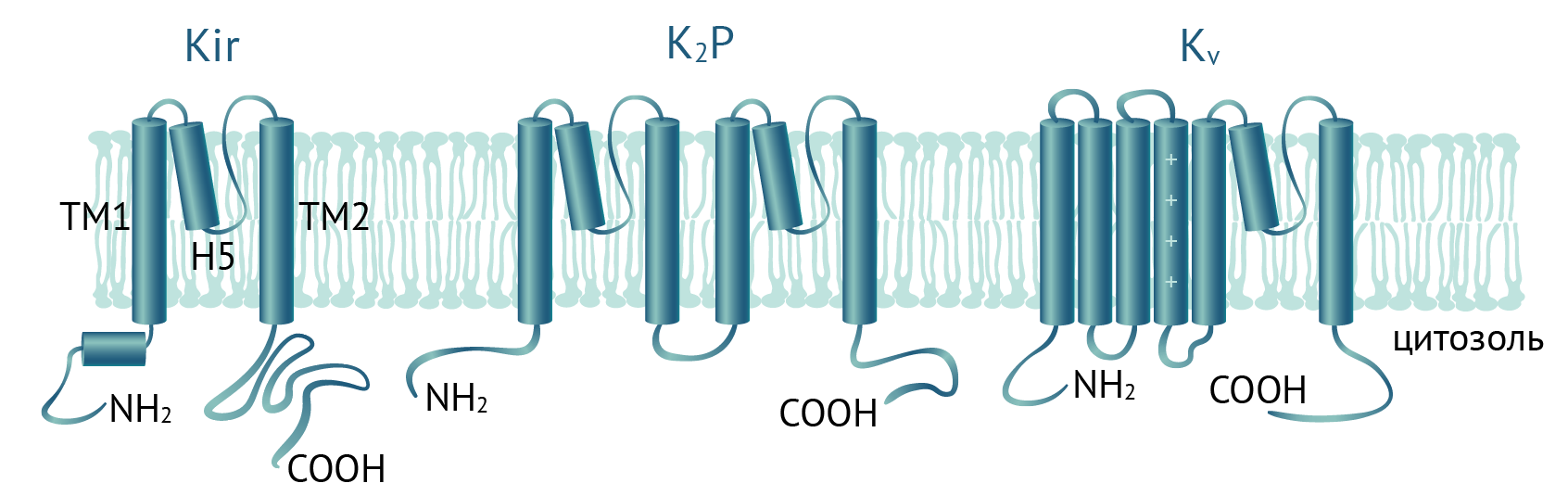
Рисунок 2
Доменная организация калиевых каналов различных семейств: калиевых каналов внутреннего выпрямления (Kir), калиевых каналов с двумя поровыми доменами (K2P) и потенциалзависимых калиевых каналов (Kv). Субъединица Kir состоит из двух альфа-спиралей, пересекающих мембрану, NH2- и COOH- концы обращены в цитозоль. Между трансмембранными спиралями находится петля, образующая селективный фильтр канала. Селективный фильтр всех калиевых каналов устроен по единому принципу и обеспечивает их высокую избирательность.
В настоящее время известно 16 генов субъединиц Kir каналов, которые группируют в семь подсемейств (Kir1.x–Kir7.x) на основании сходства структуры. Функционально их можно разделить на четыре группы:
— классические Kir (Kir2.x);
— транспортные каналы (Kir1.x, Kir4.x, Kir5.x и Kir7.x);
— каналы, активируемые G-белками (Kir3.x), также называемые GIRK (G protein-coupled inwardly-rectifying K+ channels);
— АТФ-чувствительные K+-каналы (Kir6.x) — KATP каналы.
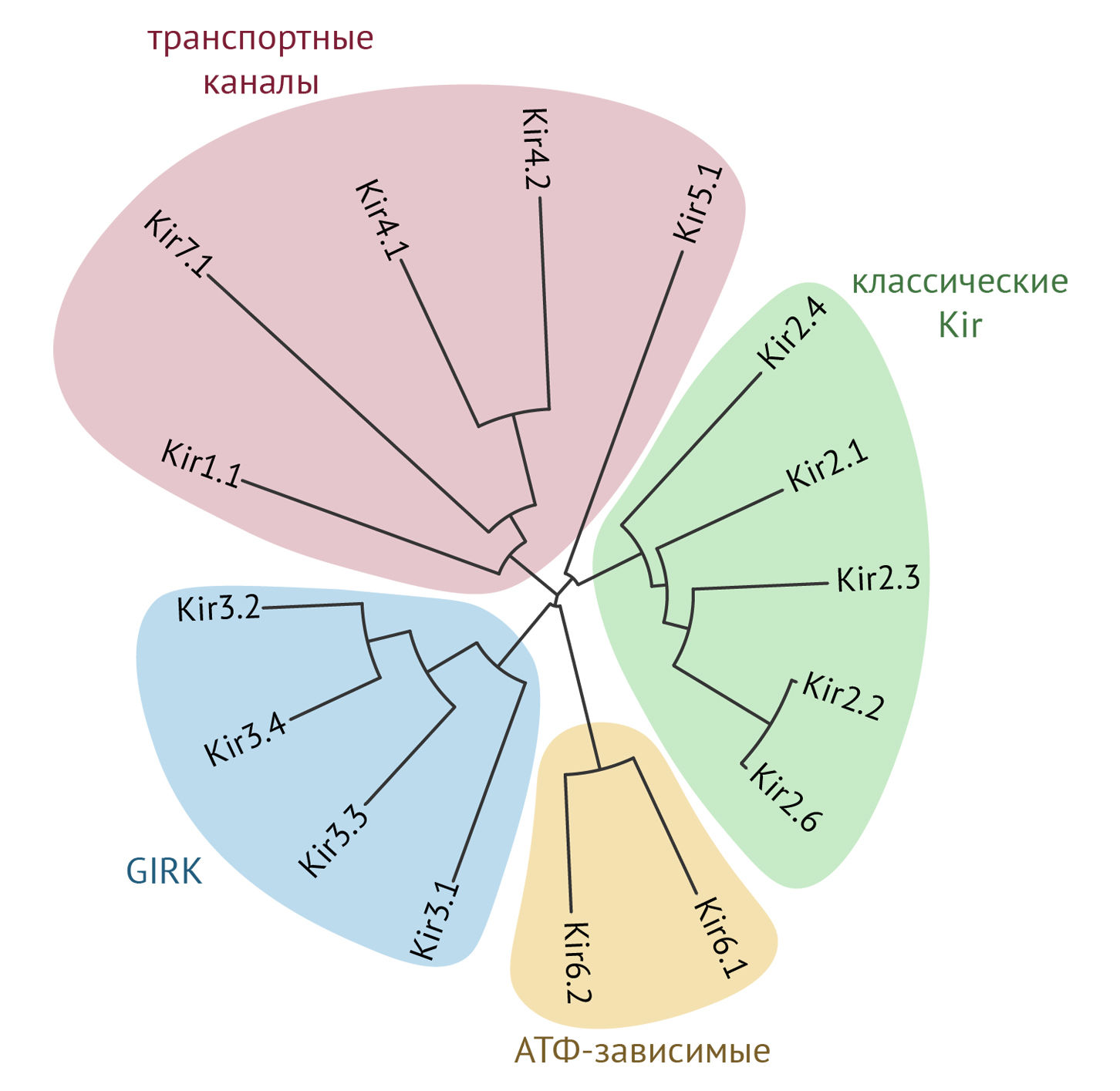
Рисунок 3
Филогенетическое древо аминокислотных последовательностей калиевых каналов внутреннего выпрямления.
Каналы семейства Kir могут образовывать как гомомеры, так и гетеромеры из четырех субъединиц, обычно в рамках одного подсемейства (за исключением Kir4.1/Kir5.1). Гетеромеризация влияет на электрофизиологические и фармакологические свойства каналов, а также на их внутриклеточную локализацию, тем самым расширяя функциональные возможности.
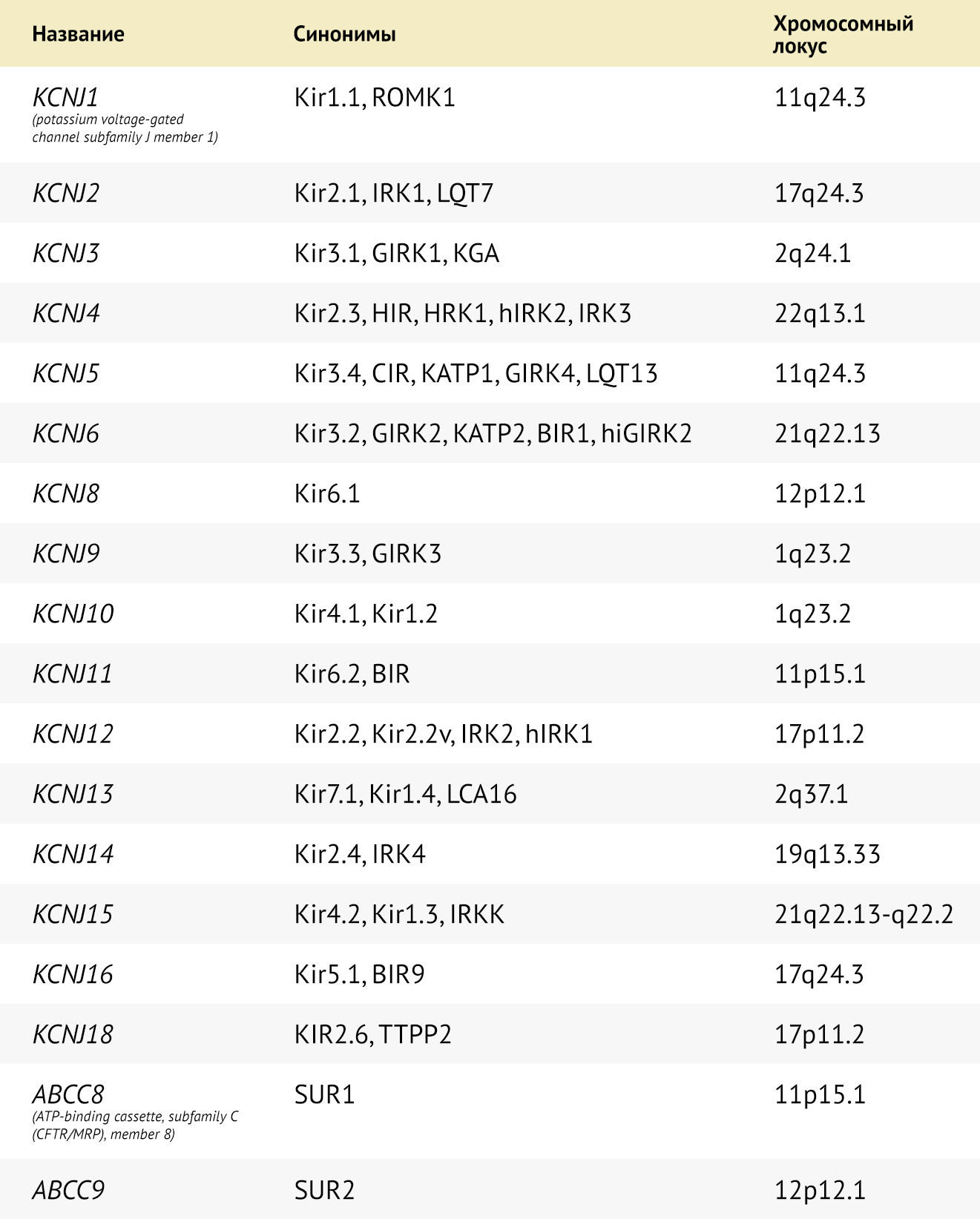
Таблица 1
Семейство генов калиевых каналов внутреннего выпрямления [6].
Механизмы работы и регуляции каналов Kir
Внеклеточная концентрация K+ и Na+
При увеличении внеклеточной концентрации калия проводимость большинства калиевых каналов внутреннего выпрямления (кроме Kir7.1 [7]) квадратично возрастает [2, 8]. Такое поведение обусловливает парадоксальную гиперполяризацию мембраны гладкомышечных клеток в ответ на умеренное повышение уровня K+. Однако в исследовании 2018 года предложено иное объяснение этому эффекту: активность канала зависит не от концентрации калия, а от конкуренции между внеклеточным калием и натрием, который блокирует пору канала снаружи [9].
Внутриклеточные поливалентные катионы
Внутреннее выпрямление — отличительная характеристика каналов Kir — обусловлено блоком выходящего тока калия внутриклеточными дивалентными катионами, такими как Mg2+ [10], и полиаминами (спермин, спермидин, путресцин и кадаверин) [11].
Когда мембрана гиперполяризована, ионы K+ свободно проходят через пору канала. Однако при деполяризации мембраны катионы под действием электрического поля поступают из цитоплазмы в пору и блокируют проход для ионов K+. Этот механизм позволяет каналам Kir поддерживать потенциал покоя и не рассеивать электрохимический градиент при генерации потенциала действия.
В случае возникновения гиперполяризации снятие блокировки канала происходит в две ступени: сначала диссоциирует ион Mg2+, а через некоторое время благодаря диссоциации полиаминов выпрямление тока усиливается.
Степень выраженности внутреннего выпрямления различается для разных подсемейств Kir. По характеру выпрямления выделяют «сильно-» (Kir2.x и Kir3.x), «средне-» (Kir4.x) и «слабовыпрямляющие» (Kir1.1 и Kir6.x) каналы. В основе молекулярного механизма блока канала внутриклеточными поливалентными катионами лежат несколько отрицательно заряженных аминокислотных остатков в цитоплазматической части поры канала. Остаток аспартата Asp172 в Kir2.1 отвечает за высокую аффинность этого канала к магнию и сильное выпрямление, тогда как в Kir1.1 соответствующий остаток аспарагина Asn171 не заряжен. Мутация Asn171Asp (нейтральная аминокислота заменяется на кислую) увеличивает аффинность Kir1.1 к Mg2+ и, соответственно, внутреннее выпрямление этого канала [12]. Другие отрицательно заряженные остатки в цитоплазматической части поры и вестибулуме канала отвечают за медленный блок полиаминами [13].
Фосфатидилинозитол-4,5-бисфосфат (PIP2)
Долгое время мембранные фосфолипиды считались инертной средой, в которую погружены ионные каналы, однако за последние десятилетия стало ясно, что фосфолипиды активно участвуют в регуляции активности ионных каналов [14].
Анионный фосфолипид плазматической мембраны фосфатидилинозитол-4,5-бисфосфат (PtdIns(4,5)P2, или PIP2) необходим для нормальной работы большинства каналов Kir [15–17]. PIP2 связывается с положительно заряженными аминокислотными остатками в гибком участке, связывающем C-концевой (цитозольный) и трансмембранный домены молекулы канала, и таким образом активирует канал [18]. В этой роли мембранный фосфолипид PIP2 принципиально не отличается от растворимых лигандов, активирующих другие ионные каналы, значит, каналы семейства Kir можно считать лиганд-управляемыми [14]. Мутации, нарушающие это взаимодействие, могут быть причиной некоторых заболеваний человека [19]. Например, мутации Kir2.1 в позиции Arg218 (Arg218Gln/Trp) связаны с синдромом Андерсена — Тавила (периодический паралич мускулатуры, удлинение интервала QT и характерные физические особенности) [20], а мутации Kir1.1 в позиции Arg311 (Arg311Gln/Trp) вызывают неонатальный синдром Барттера [21], сопровождающийся излишней экскрецией солей, гипокалиемическим алкалозом и пониженным артериальным давлением (см. раздел «Kir1.1 — синдром Барттера»).
Внутриклеточный и внеклеточный pH
Некоторые представители семейства Kir чувствительны к изменениям кислотности внутри- или внеклеточной среды. Kir1.1 закрываются при закислении цитозоля, а мутации, сдвигающие pKa к более щелочным значениям, связаны с синдромом Барттера. Гетеромеры Kir4.1/5.1 также закрываются при снижении внутриклеточного pH. Гетеромеры экспрессируются в pH-чувствительных нейронах голубого пятна и, вероятно, опосредуют реакцию на гиперкапнию (см. раздел «Kir4.1 и Kir5.1 в нервной системе»).
Kir2.x — классические калиевые каналы внутреннего выпрямления
Калиевые каналы внутреннего выпрямления в скелетных мышцах и миокарде принадлежат к подсемейству Kir2.x. Каналы этого подсемейства постоянно открыты и обладают сильным внутренним выпрямлением, благодаря чему они поддерживают очень высокий потенциал покоя и долгую фазу плато потенциала действия в кардиомиоцитах и некоторых других типах клеток (см. рис. 4).
К этому подсемейству относятся:
— IRK1/Kir2.1/KCNJ2 [2];
— Kir2.2 (IRK2)/KCNJ12 [22,23];
— Kir2.3 (IRK3, BIR11, HIR)/KCNJ4 [24–26];
— нейрональный Kir2.4 (IRK4)/KCNJ14 [27];
— Kir2.6/KCNJ18 [28], который более, чем на 98 % совпадает с Kir2.2.
Kir2.1, Kir2.2 и Kir2.3 могут образовывать гомо- и гетеротетрамеры в любых комбинациях [29]. Некоторые гетеромерные комбинации встречаются in vivo, например, Kir2.1/2.2 и Kir2.1/2.3 в кардиомиоцитах и Kir2.1/2.4 в мозге [30]. Kir2.6 гетеромеризуется с Kir2.1 и Kir2.2 и ограничивает экспрессию этих каналов на мембране клетки, задерживая их в ЭПР [31].
Для нормальной активности каналов Kir2.x необходим PIP2. Kir2.3 активируется повышением внутри- или внеклеточного pH, за чувствительность к кислотности среды отвечает единственный остаток His117 [32, 33], Kir2.4 также имеет остаток гистидина His130, соответствующий His117 в Kir2.3, и благодаря этому активируется повышением внеклеточного pH.
Физиологическая роль
Kir2.x в сердце
Классические Kir-каналы экспрессируются в кардиомиоцитах предсердий [34], желудочков [35, 36] и волокон Пуркинье [37, 38], но не в клетках пейсмейкеров [39]. Эти каналы пропускают высокоамплитудный входящий ток при Em, более отрицательном, чем EK, и относительно большой выходящий ток IK1 при Em чуть ниже EK, но при удалении значения мембранного потенциала клетки от равновесного потенциала калия ток быстро ослабевает. Такое поведение классического тока внутреннего выпрямления IK1 стабилизирует потенциал покоя кардиомиоцитов. Каналы Kir2.x также принимают участие в поддержании деполяризации в фазу плато потенциала действия кардиомиоцитов. При положительных потенциалах IK1 практически равен нулю, что не позволяет K+ преждевременно выходить из клетки и вызывать реполяризацию мембраны. Однако, когда активация потенциалзависимых калиевых каналов запускает реполяризацию и потенциал достигает значений, при которых ток внутреннего выпрямления растет, а не уменьшается при приближении к EK, высокоамплитудный IK1 ускоряет финальную фазу реполяризации и возвращает мембранный потенциал к потенциалу покоя.
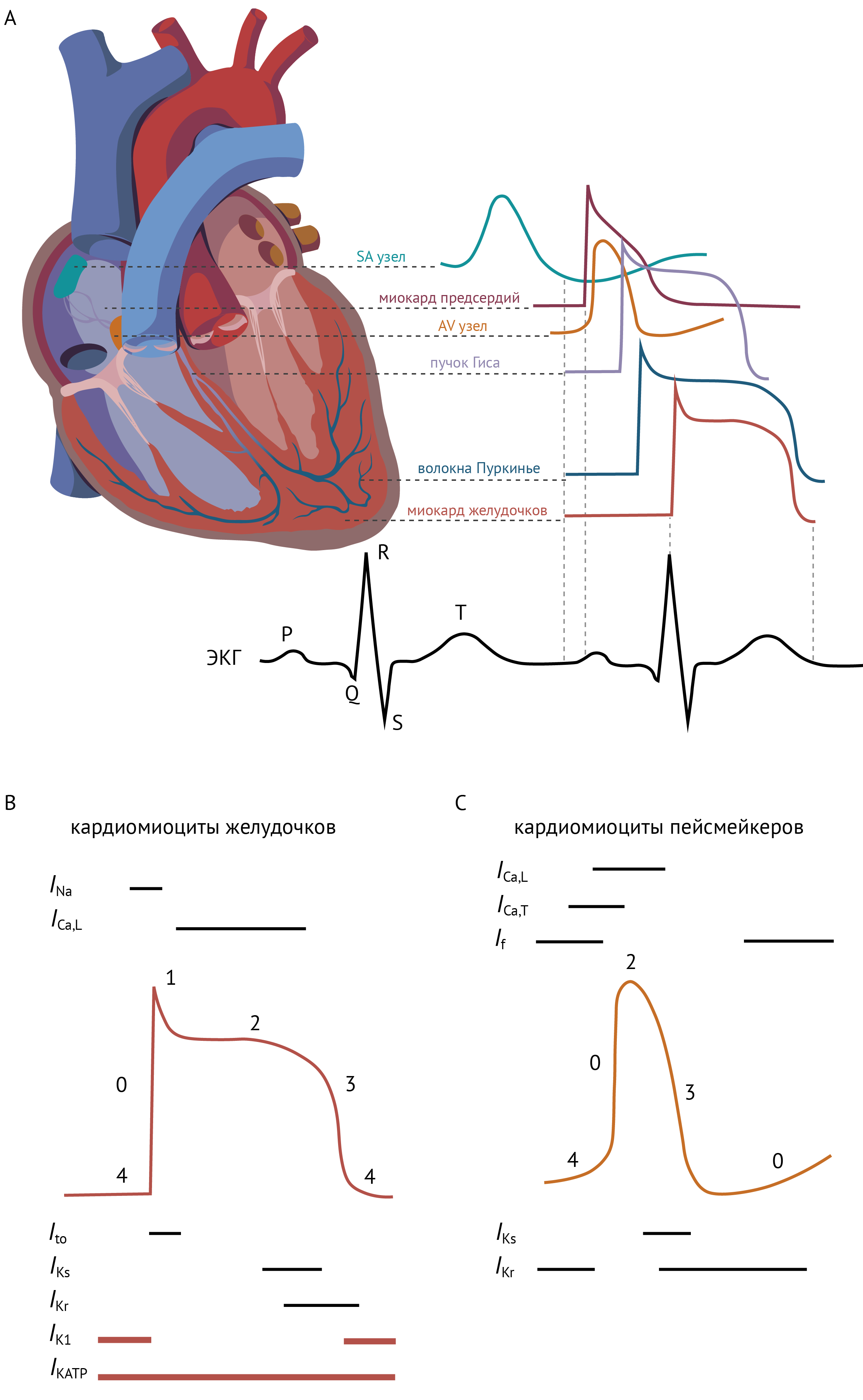
Рисунок 4. Потенциалы действия в сердце [40]
А. Потенциалы действия в различных отделах сердца. B и C. Токи, участвующие в формировании потенциала действия в кардиомиоцитах желудочков (B) и пейсмейкеров (C).
0 — фаза быстрой деполяризации; 1 — фаза быстрой начальной реполяризации; 2 — фаза плато; 3 — фаза окончательной реполяризации; 4 — потенциал покоя.
Входящие токи (только ?-субъединицы каналов): INa опосредован Nav1.5; ICa,L — Cav1.2; ICa,T — Cav3.x; If — HCN (управляемые циклическими нуклеотидами активируемые гиперполяризацией каналы);
Выходящие токи: Ito — Kv4.3; быстро активирующийся IKr — Kv11.1; медленно активирующийся IKs — Kv7.1, IK1 — Kir2.1; IKATP — Kir6.2/SUR2A. Классические каналы Kir отсутствуют в кардиомиоцитах пейсмейкеров, из-за чего мембранный потенциал покоя в них нестабилен.
Отсутствие классических Kir каналов в клетках пейсмейкеров определяет низкий и нестабильный потенциал покоя этих клеток, а также отсутствие фазы плато в потенциале действия.
Какие именно изоформы Kir2.x опосредуют IK1? В сердце экспрессируются Kir2.1, Kir2.2 и Kir2.3 [5]. Исследования на нокаутных мышах показали, что нокаут Kir2.2 ведет к уменьшению амплитуды тока на 50 %, тогда как в кардиомиоцитах нокаута Kir2.1 IK1 отсутствовал [41]. Из этого следует, что in vivo Kir2.2 образуют гетеромеры с Kir2.1. Экспрессия доминантно негативной формы Kir2.1 или Kir2.2 в желудочковых миоцитах кролика приводила к уменьшению IK1 на 70 %, что также свидетельствует о гетеромерных комплексах Kir2.1/Kir2.2 [42].
Фенотип нокаутных мышей, не экспрессирующих Kir2.1, характеризуется нестабильным потенциалом покоя и ритмичными потенциалами действия в желудочковых кардиомиоцитах. Потенциалы действия этих кардиомиоцитов были значительно длиннее, чем у кардиомиоцитов дикого типа. При этом у Kir2.1-нокаутных мышей на ЭКГ отсутствуют эктопические систолы и аритмии обратного входа импульса (англ. re-entry), что свидетельствует о нормальной работе синусового узла. Но, несмотря на это, Kir2.1-нокауты умирают уже через 12 часов после рождения из-за нарушений дыхания, так как полная расщелина твердого неба не позволяет этим мышам нормально питаться и содержимое полости рта попадает в трахею, вызывая асфиксию [43], что свидетельствует о важной роли этих каналов в эмбриогенезе.
Kir2.x в кровеносных сосудах
Классические каналы Kir экспрессируются в гладкомышечных клетках и в эндотелии сосудов [44, 45] и участвуют в поддержании сосудистого тонуса.
В клетках гладких мышц резистивных артерий классические Kir-каналы способствуют вазодилатации в ответ на увеличение [K+]o (см. рис. 6). Этот механизм особенно важен для мозговых и коронарных артерий. Хотя повышение [K+]o обычно приводит к деполяризации и, следовательно, констрикции гладкомышечных клеток, небольшое увеличение [K+]o от 6 до 15 мМ гиперполяризует клетку и ведет к расширению мозговых и коронарных артерий. [46–48].
Потенциал покоя гладкомышечных клеток артерий находится на уровне около –45 мВ, а рост [K+]o повышает мембранный потенциал до приблизительно –60 мВ вследствие увеличения проводимости Kir-каналов [46, 48]. При гиперполяризации мембраны потенциал-зависимые кальциевые каналы закрываются и внутриклеточная концентрация Ca2+ уменьшается, что ведет к вазодилатации [49].
Вокруг гладкомышечных клеток мозговых артерий концентрация K+ повышена из-за секреции калия пластинчатыми окончаниями отростков астроцитов. Это локальное увеличение [K+]o происходит во время стимуляции нейронов, таким образом, этот механизм участвует в сопряжении активности нейронов и регуляции локального кровотока в мозге [50].
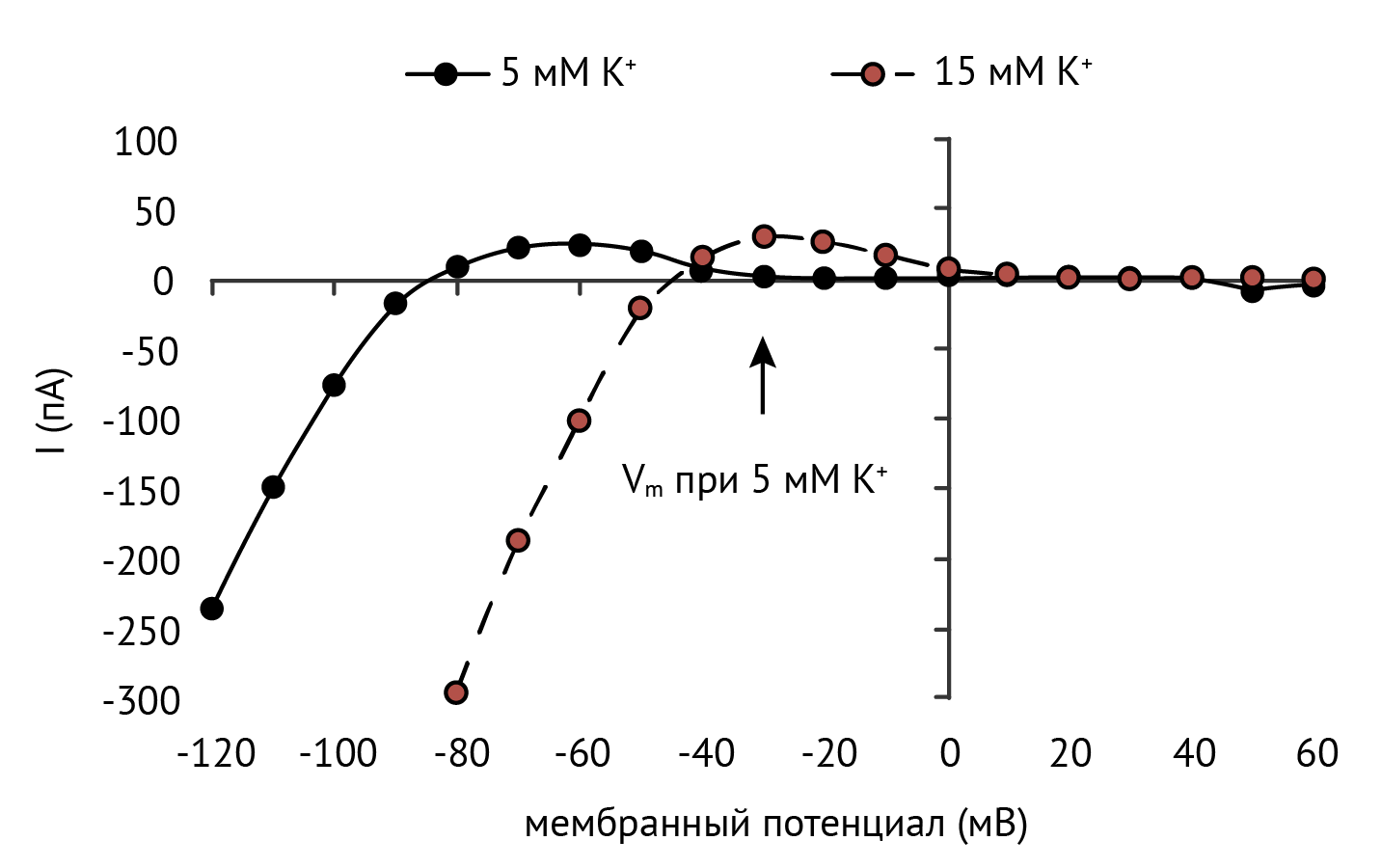
Рисунок 5
IK1 в при 5 и 15 мМ [K+]o. Повышение [K+]o приводит к усилению IK1 и сдвигу потенциала покоя в сторону EK (пояснения в тексте) [51].
Kir2.1 играет важную роль в вазодилатации в ответ на повышение [K+]o в гладкомышечных клетках. В пользу этого свидетельствуют измерения уровня транскриптов Kir2.x в гладкомышечных клетках сосудов, в которых присутствует Kir2.1, а Kir2.2 и Kir2.3 обнаружить не удалось [52], причем уровень экспрессии Kir2.1 и плотность K+ тока внутреннего выпрямления увеличивается с движением дистально по сосудистому древу от артерий эластического типа к резистивным артериям [53], которые участвуют в дифференциальной регуляции кровотока различных органов. Кроме того, мозговые артерии Kir2.2-нокаутных мышей, в отличие от сосудов мышей, нокаутных по Kir2.1, нормально расширяются в ответ на повышение внеклеточной концентрации калия [43].
Kir2.1 также участвуют в эндотелий-зависимой вазодилатации. Один из механизмов, показанных для капилляров и артериол мозга, аналогичен гладкомышечному ответу на повышение [K+]o. В эндотелиальных клетках капилляров мозга, в отличие от эндотелия артериол, отсутствуют кальций-зависимые калиевые каналы малой (обозначаются SK — small conductance) и промежуточной проводимости (IK — intermediate conductance), а Kir2.1 является главным медиатором калиевого тока. В покое клетки эндотелия деполяризованы (40 мВ), а EK составляет для них –103 мВ (в цереброспинальной жидкости). Нейроны при генерации потенциалов действия выделяют K+, который проходит через астроциты к капиллярам (подробнее о механизмах транспорта K+ астроцитами см. в разделе «Kir4.1 и Kir5.1 в нервной системе») и активирует ток через Kir2.1-каналы на эндотелии. Увеличение проводимости мембраны для K+ приближает мембранный потенциал к новому EK, который будет сдвинут вправо из-за повышения внеклеточного содержания K+ (см. рис. 5). Гиперполяризация мембраны эндотелия капилляров может распространяться через щелевые контакты вверх по сосудистому древу к артериолам, вызывая их расширение [54], таким образом кровоток может подстраиваться под текущую потребность ткани (функциональная гиперемия).
Связь между активностью нейронов и кровотоком лежит в основе метода функциональной магнитно-резонансной томографии, и описанный механизм эндотелий-зависимой вазодилатации участвует в генерации BOLD (blood oxygenation level-dependent — зависящего от насыщения крови кислородом) сигнала [55, 56]. Роль классических Kir каналов в функциональной гиперемии скелетных мышц и сердца в настоящее время не изучена.
Существуют данные об участии Kir2.x эндотелия артериол в вазодилатации, однако детали этого процесса не ясны. Стимуляция мускариновых рецепторов на клетках эндотелия приводит к повышению цитозольной концентрации Ca2+ через активацию IP3R на ЭПР и активацию кальциевых каналов TRPV4 на плазматической мембране, вызванную фосфорилированием PKC. Повышение [Ca2+]i с одной стороны, стимулирует продукцию эндотелиальных гиперполяризующих факторов (PGI2 и другие производные арахидоновой кислоты и NO), а, с другой стороны, приводит к активации SK и IK кальций-зависимых калиевых каналов и гиперполяризации эндотелиальных клеток. Ток через Kir2.1 усиливается при гиперполяризации мембраны и способствует дальнейшей гиперполяризации, которая затем распространяется через щелевые контакты на гладкомышечные клетки. Распространение гиперполяризации и вазодилатация чувствительны к блокатору Kir каналов Ba2+, а у мышей с эндотелий-специфичным нокаутом KCNJ2 была нарушена вазодилатация в мелких брыжеечных артериях [57]. Однако в других исследованиях получены результаты, не согласующиеся с этой моделью. Так, Smith и соавторы не обнаружили экспрессии Kir2.x в брыжеечных артериях крыс и разницы в ответе артерий на аппликацию Ba2+ [58].
Некоторые работы связывают роль Kir2.x каналов в вазодилатации в ответ на напряжение сдвига с продукцией NO: так, блок Kir2.x каналов Ba2+ предотвращает Ca2+-зависимую продукцию NO и опосредованную им вазодилатацию [59], влияя на уровень фосфорилирования эндотелиальной NO-синтазы eNOS [60], однако события, связывающие активацию каналов Kir и фосфорилирование eNOS, неизвестны. В других работах оспаривается необходимость изменения фосфорилирования eNOS для вазодилатации в ответ на ток крови в случае предварительной констрикции артериол [61].
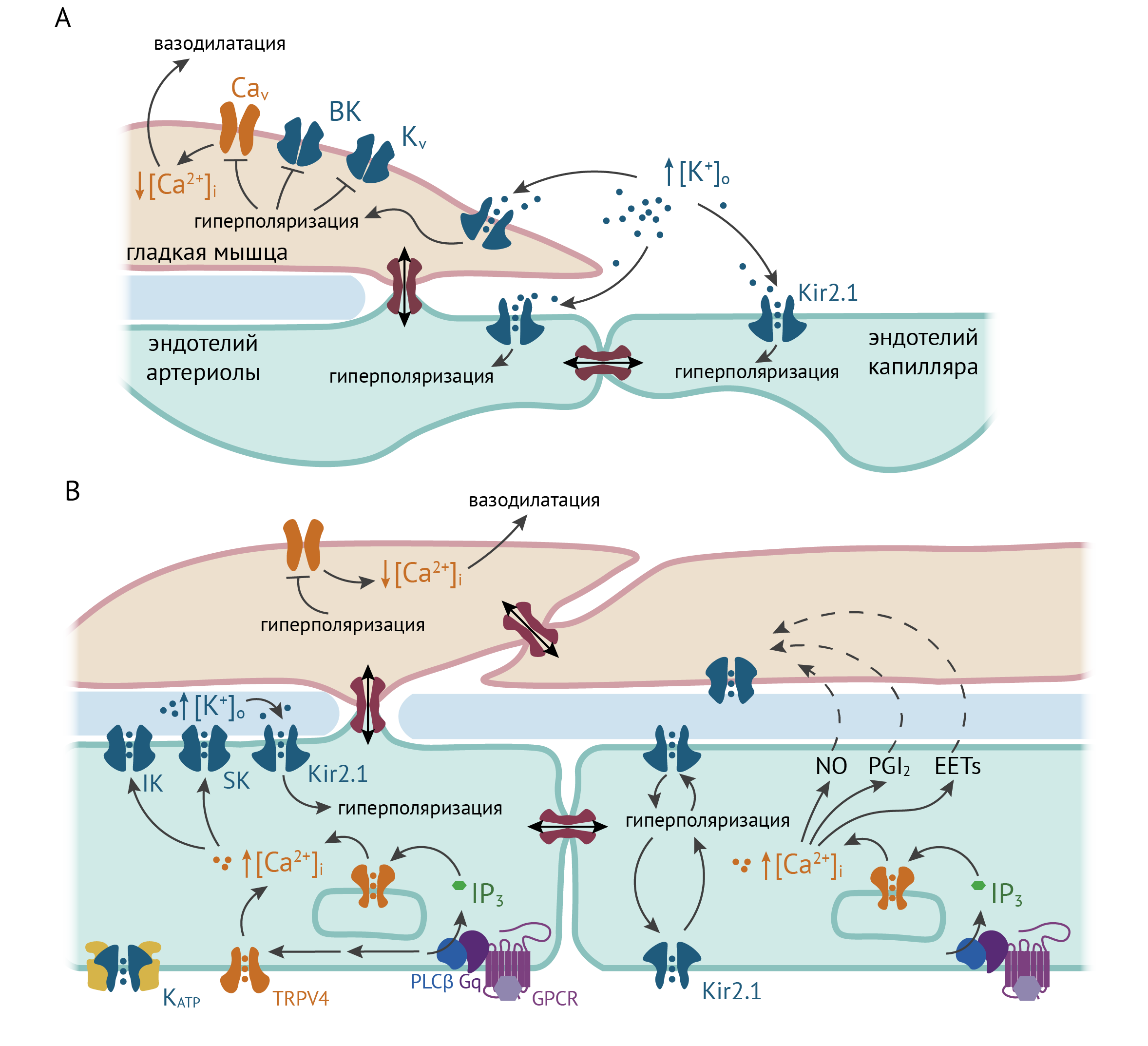
Рисунок 6. Роль Kir2.1 в гладкомышечных клетках и эндотелии кровеносных сосудов
A. Активация Kir2.1 на клетках эндотелия и гладких мышц в ответ на повышение [K+]o способствует гиперполяризации мембраны, распространяющейся по щелевым контактам, в том числе и вверх по сосудистому древу [54]. B. Некоторые механизмы эндотелий-зависимой вазодилатации. Повышение [Ca2+]i стимулирует продукцию эндотелиальных гиперполяризующих факторов (NO, PGI2, EETs — эпоксиэйкозатриеновые кислоты). Повышение [Ca2+] активирует кальций-зависимые калиевые каналы IK, SK, а Kir2.1 усиливают гиперполяризацию.
Мутации в Kir2.x
Синдром Андерсена — Тавила (LQT7)
Мутации утраты функции в Kir2.1-каналах ведут к синдрому Андерсена — Тавила (LQT7) [20], который наследуется по аутосомно-доминантному типу или возникает de novo. Некоторые мутации нарушают взаимодействия с PIP2, а некоторые — доставку канала на плазматическую мембрану. Для этого синдрома характерны следующие признаки: эпизоды периодического паралича, нарушение сердечного ритма (желудочковые аритмии и удлинение интервала QT) и признаки дисморфогенеза, однако полная триада симптомов встречается редко [62]. Особенности строения лица пациентов с синдромом Андерсена — Тавила и расщелина неба у нокаутных мышей возникают вследствие нарушения сигнального каскада BMP (bone morphogenic protein) [63, 64]. На рисунке 7 изображен пациент с типичными для этого синдрома чертами лица.
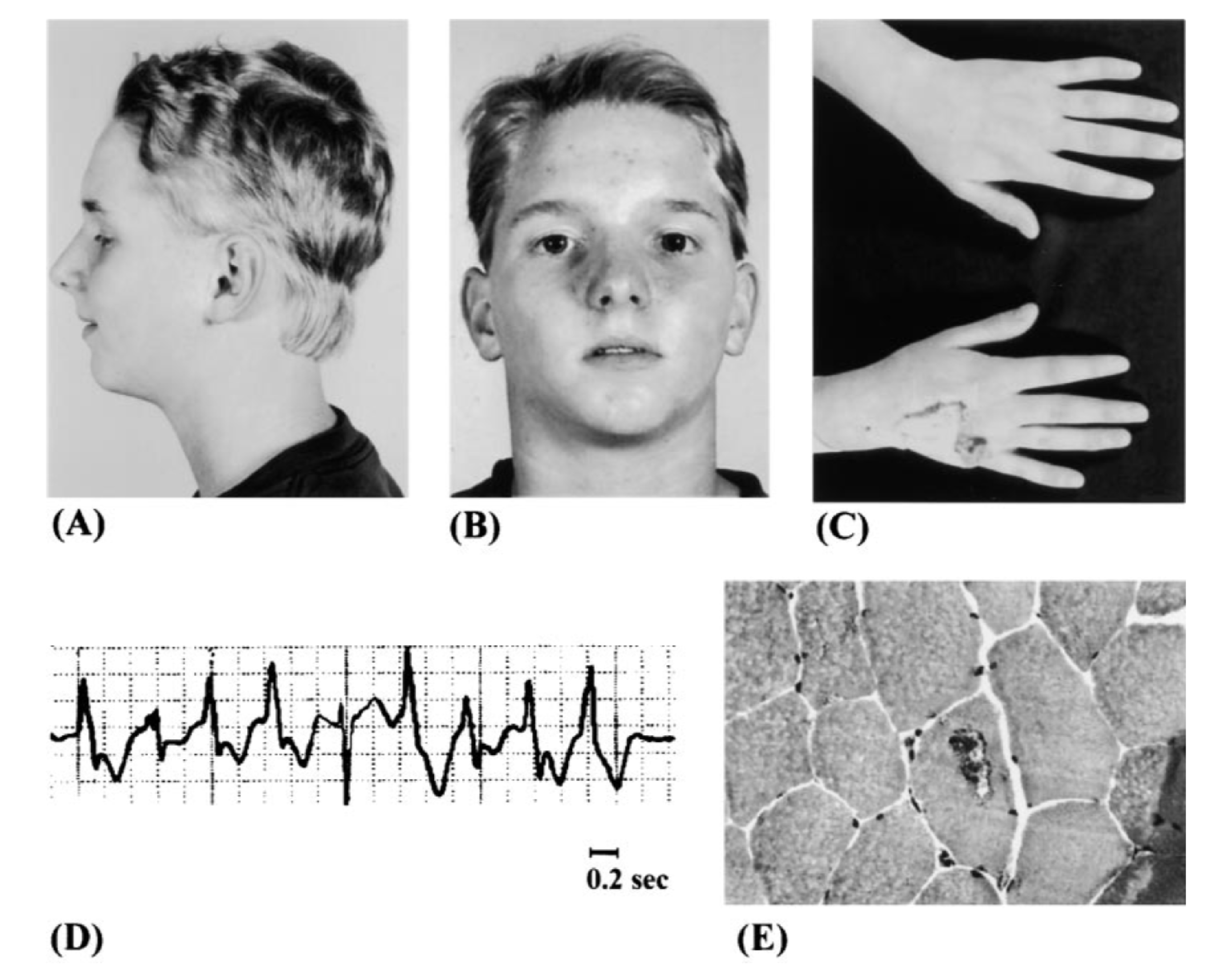
Рисунок 7. Синдром Андерсена — Тавила
A, B, С. Типичные черты лица при синдроме Андерсена — Тавила: низко посаженные уши, гипертелоризм, микрогнатия и клинодактилия. D. Полиморфная желудочковая тахикардия на ЭКГ. E. Тубулярные агрегаты на биопсии мышцы [20].

Рисунок 8. Синдром удлиненного интервала QT
Синдром короткого интервала QT (SQT3)
Мутации усиления функции в KCNJ2 могут приводить к синдрому короткого интервала QT (SQT3) [66, 67]. Усиление поступления K+ при положительных потенциалах ускоряет реполяризацию, что на ЭКГ выражается в укорочении интервала QT. Морфология сердца при этом остается нормальной, но повышен риск возникновения фибрилляции предсердий и желудочков и внезапной смерти [40, 68].
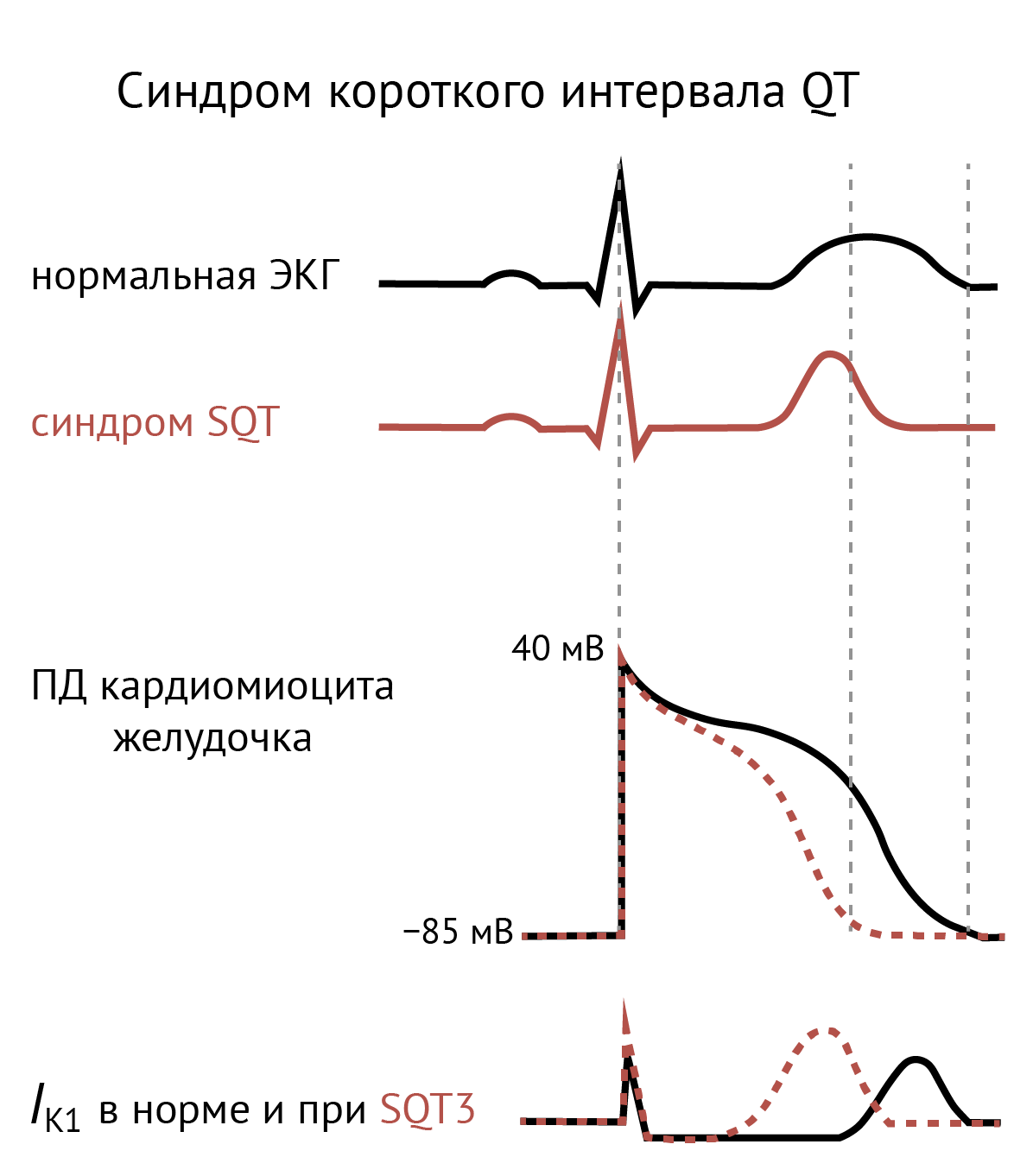
Рисунок 9. Синдром короткого интервала QT
Тиреотоксический периодический паралич
Мутации утраты функции в гене KCNJ18, кодирующем Kir2.6, ведут к тиреотоксическому периодическому параличу [28]. Этот ген экспрессируется в скелетных мышцах, и его транскрипция находится под контролем тиреоидных гормонов. Пациенты испытывают эпизоды паралича и гипокалиемии, причем между этими приступами никакие симптомы не проявляются. Эпизоды прекращаются при компенсации гипертиреоза.
Предполагаемый механизм развития слабости или паралича (рис. 10) включает в себя связывание комплекса рецептора тиреоидных гормонов TR? и T3 на промоторе гена KCNJ18 c элементом, чувствительным к тиреоидным гормонам, что приводит к повышению экспрессии этого рецептора. Если повышается экспрессия мутантного варианта с утраченной функцией, амплитуда IK снижается, мембрана деполяризуется и потенциалзависимые натриевые и кальциевые каналы инактивируются. Это приводит к изменению возбудимости мышечных волокон и слабости/параличу. Кроме того, в состоянии тиреотоксикоза повышается активность PKC и снижается уровень PIP2, что, в свою очередь, также приводит к снижению калиевого тока внутреннего выпрямления.
Интересно, что часть мутаций, выявленных у пациентов с тиреотоксическим периодическим параличом, являются мутациями усиления, а не утраты функции. Так, мутация T354M приводит к незначительному снижению тока в состоянии покоя, однако каналы с этой мутацией не чувствительны к PKC, которая в норме фосфорилирует канал и снижает вероятность его открытия. В присутствии этой мутации канал остается открытым, и при высокой активности PKC мембрана гиперполяризуется и возбудимость мышечного волокна уменьшается.
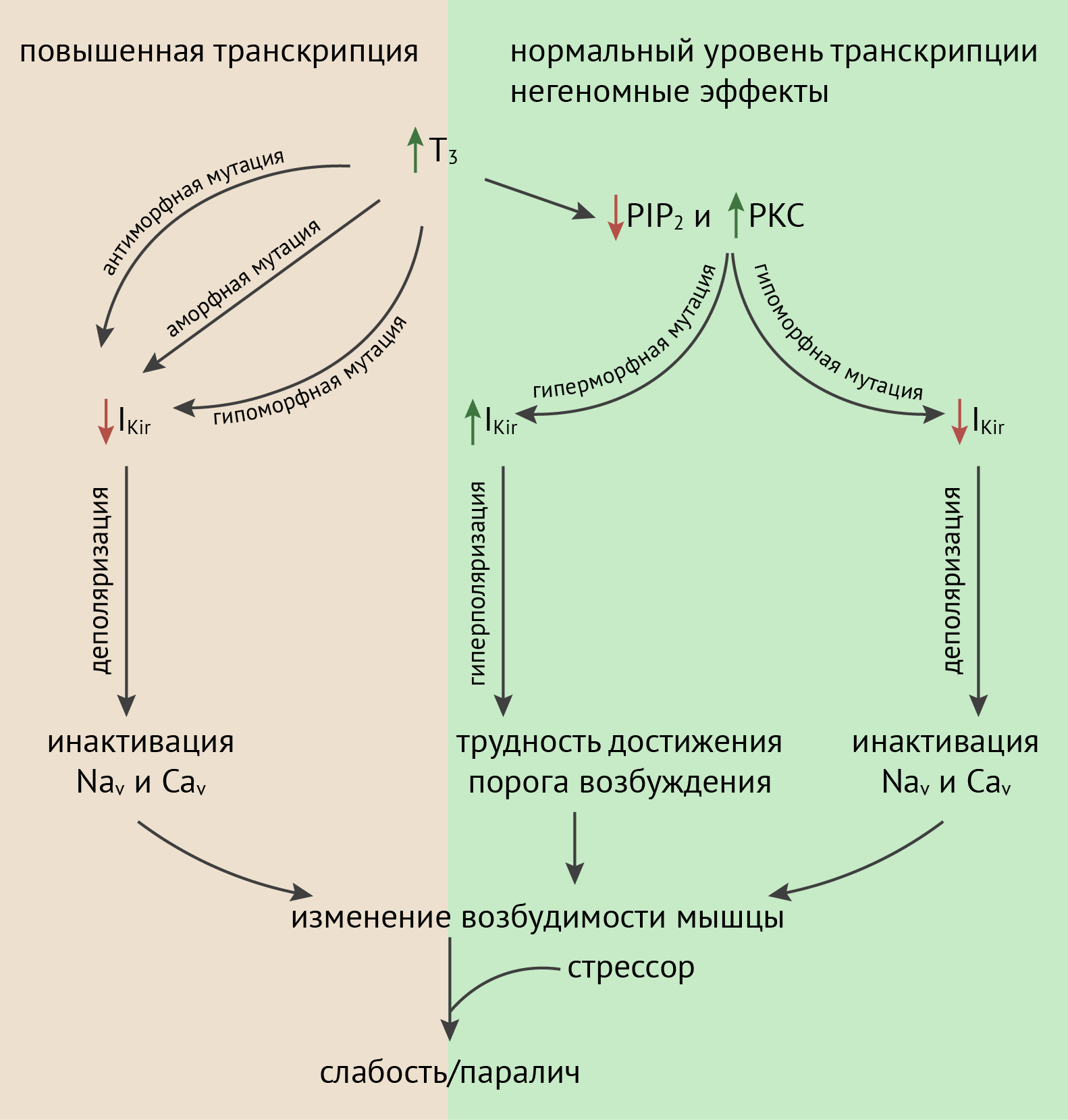
Рисунок 10. Механизм развития периодического паралича при тиреотоксикозе [28]
В последующем исследовании группа ученых изучила популяции из Китая с болезнью Грейвса и обнаружила и другие варианты, связанные с тиреотоксическим периодическим параличом, в некодирующей области на хромосомном локусе 17q24.3, в котором располагаются гены KCNJ2 и KCNJ16. Поскольку экспрессия KCNJ16 (Kir5.1) отсутствует в скелетных мышцах, они посчитали KCNJ2 более подходящим кандидатом [69]. Похожие результаты дало исследование популяции из Таиланда [70].
В следующих частях будут рассмотрены транспортные, активируемые G-белками и АТФ-зависимые калиевые каналы внутреннего выпрямления, а также их фармакологические свойства.
Источники:
- Katz B. Les constantes electriques de la membrane du muscle // Arch. Sci. Phisiol. 1949. Vol. 3. P. 285–299.
- Kubo Y. et al. Primary structure and functional expression of a mouse inward rectifier potassium channel // Nature. 1993. Vol. 362, № 6416. P. 127–133.
- Ho K. et al. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel // Nature. 1993. Vol. 362, № 6415. P. 31–38.
- Kubo Y. et al. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel // Nature. 1993. Vol. 364, № 6440. P. 802–806.
- Hibino H. et al. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles // Physiol. Rev. 2010. Vol. 90, № 1. P. 291–366.
- Potassium voltage-gated channel subfamily J (KCNJ) Gene Family | HUGO Gene Nomenclature Committee [Electronic resource]. URL: https://www.genenames.org/cgi-... (accessed: 04.08.2018).
- D?ring F. et al. The epithelial inward rectifier channel Kir7.1 displays unusual K+ permeation properties // J. Neurosci. 1998. Vol. 18, № 21. P. 8625–8636.
- Lopatin A.N., Nichols C.G. [K+] dependence of open-channel conductance in cloned inward rectifier potassium channels (IRK1, Kir2.1) // Biophys. J. Elsevier, 1996. Vol. 71, № 2. P. 682–694.
- Ishihara K. External K+ dependence of strong inward rectifier K+ channel conductance is caused not by K+ but by competitive pore blockade by external Na+ // J. Gen. Physiol. 2018. Vol. 150, № 7. P. 1–14.
- Matsuda H., Saigusa A., Irisawa H. Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+ // Nature. 1987. Vol. 325, № 6100. P. 156–159.
- Lopatin A.N., Makhina E.N., Nichols C.G. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification // Nature. 1994. Vol. 372, № 6504. P. 66–369.
- Lu Z., MacKinnon R. Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel // Nature. 1994. Vol. 371, № 6494. P. 243–246.
- Kurata H.T. et al. The Role of the Cytoplasmic Pore in Inward Rectification of Kir2.1 Channels // J. Gen. Physiol. 2007. Vol. 130, № 2. P. 145–155.
- Hansen S.B. Lipid agonism: The PIP2 paradigm of ligand-gated ion channels // Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. Elsevier B.V., 2015. Vol. 1851, № 5. P. 620–628.
- Hilgemann D.W., Ball R. Regulation of cardiac Na+, Ca2+ exchange and KATP Potassium channels by PIP2 // Science. 1996. Vol. 273, № 5277. P. 956–959.
- Huang C.L., Feng S., Hilgemann D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by G?? // Nature. 1998. Vol. 391, № 6669. P. 803–806.
- Hilgemann D.W., Feng S., Nasuhoglu C. The Complex and Intriguing Lives of PIP2 with Ion Channels and Transporters // Sci. Signal. 2003. Vol. 2001, № 111. P. re19–re19.
- Hansen S.B., Tao X., MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2 // Nature. 2011. Vol. 477, № 7365. P. 495–498.
- Lopes C.M.B. et al. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies // Neuron. 2002. Vol. 34, № 6. P. 933–944.
- Plaster N.M. et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome // Cell. 2001. Vol. 105, № 4. P. 511–519.
- Schulte U. et al. pH gating of ROMK (Kir1.1) channels: control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome // Proc. Natl. Acad. Sci. 1999. Vol. 96, № 26. P. 15298–15303.
- Koyama H. et al. Molecular cloning, functional expression and localization of a novel inward rectifier potassium channel in the rat brain // FEBS Lett. 1994. Vol. 341, № 2–3. P. 303–307.
- Fan Z., Makielski J.C. Anionic phospholipids activate ATP-sensitive potassium channels // J. Biol. Chem. 1997. Vol. 272, № 9. P. 5388–5395.
- Bredt D.S. et al. Cloning and expression of two brain-specific inwardly rectifying potassium channels. // Proc. Natl. Acad. Sci. 1995. Vol. 92, № 15. P. 6753–6757.
- Morishige K. et al. Molecular cloning and functional expression of a novel brain-specific inward rectifier potassium channel. // FEBS Lett. 1994. Vol. 346, № 2–3. P. 251–256.
- Makhina E.N. et al. Cloning and expression of a novel human brain inward rectifier potassium channel. // J. Biol. Chem. 1994. Vol. 269, № 32. P. 20468–20474.
- T?pert C. et al. Kir2.4: a novel K+ inward rectifier channel associated with motoneurons of cranial nerve nuclei. // J. Neurosci. 1998. Vol. 18, № 11. P. 4096–4105.
- Ryan D.P. et al. Mutations in Potassium Channel Kir2.6 Cause Susceptibility to Thyrotoxic Hypokalemic Periodic Paralysis // Cell. 2010. Vol. 140, № 1. P. 88–98.
- Preisig-Muller R. et al. Heteromerization of Kir2.x potassium channels contributes to the phenotype of Andersen’s syndrome // Proc. Natl. Acad. Sci. 2002. Vol. 99, № 11. P. 7774–7779.
- Schram G. et al. Kir2.4 and Kir2.1 K+ channel subunits co-assemble: A potential new contributor to inward rectifier current heterogeneity // J. Physiol. 2002. Vol. 544, № 2. P. 337–349.
- Dassau L. et al. Kir2.6 Regulates the surface expression of Kir2.x inward rectifier potassium channels // J. Biol. Chem. 2011. Vol. 286, № 11. P. 9526–9541.
- Coulter K.L. et al. Identification and molecular localization of a pH-sensing domain for the inward rectifier potassium channel HIR // Neuron. 1995. Vol. 15, № 5. P. 1157–1168.
- Qu Z. et al. Gating of Inward Rectifier K + Channels by Proton-mediated Interactions of N- and C-terminal Domains // J. Biol. Chem. 2000. Vol. 275, № 41. P. 31573–31580.
- Rougier O., Vassort G., St?mpfli R. Voltage clamp experiments on frog atrial heart muscle fibres with the sucrose gap technique // Pfl?gers Arch. - Eur. J. Physiol. 1968. Vol. 301, № 2. P. 91–108.
- Kurachi Y. Voltage?dependent activation of the inward?rectifier potassium channel in the ventricular cell membrane of guinea?pig heart // J. Physiol. 1985. Vol. 366, № 1. P. 365–385.
- Beeler Jr. G.W., Reuter H. Voltage clamp experiments on ventricular myocardial fibres // J. Physiol. 1970. P. 165–190.
- McAllister R.E., Noble D. The time and voltage dependence of the slow outward current in cardiac Purkinje fibres // J. Physiol. 1966. Vol. 186, № 3. P. 632–662.
- Noble D. Electrical properties of cardiac muscle attributable to inward going (anomalous) rectification // J. Cell. Comp. Physiol. 1965. Vol. 66, № S2. P. 127–135.
- Noma A. et al. Resting K conductances in pacemaker and non-pacemaker heart cells of the rabbit. // Jpn. J. Physiol. 1984. Vol. 34, № 2. P. 245–254.
- Giudicessi J.R., Ackerman M.J. Potassium-channel mutations and cardiac arrhythmias - Diagnosis and therapy // Nat. Rev. Cardiol. 2012. Vol. 9, № 6. P. 319–332.
- Zaritsky J.J. et al. The consequences of disrupting cardiac inwardly rectifying K+ current (IK1) as revealed by the targeted deletion of the murine Kir2.1 and Kir2.2 genes // J. Physiol. 2001. Vol. 533, № 3. P. 697–710.
- Zobel C. et al. Molecular dissection of the inward rectifier potassium current (IK1) in rabbit cardiomyocytes: Evidence for heteromeric co-assembly of Kir2.1 and Kir2.2 // J. Physiol. 2003. Vol. 550, № 2. P. 365–372.
- Zaritsky J.J. et al. Targeted Disruption of Kir2.1 and Kir2.2 Genes Reveals the Essential Role of the Inwardly Rectifying K+ Current in K+-Mediated Vasodilation // Circ. Res. 2000. Vol. 87, № 2. P. 160–166.
- Nilius B., Droogmans G. Ion channels and their functional role in vascular endothelium // Physiol. Rev. 2001. Vol. 81, № 4. P. 1415-59.
- Adams D.J., Hill M.A. Potassium channels and membrane potential in the modulation of intracellular calcium in vascular endothelial cells // Journal of Cardiovascular Electrophysiology. 2004. Vol. 15, № 5. P. 598–610.
- Knot H.J., Zimmermann P.A., Nelson M.T. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels // J. Physiol. 1996. Vol. 492, № 2. P. 419–430.
- McCarron J.G., Halpern W. Potassium dilates rat cerebral arteries by two independent mechanisms // Am. J. Physiol. Circ. Physiol. 1990. Vol. 259, № 3. P. H902–H908.
- Nelson M.T. et al. Relaxation of Arterial Smooth-Muscle by Calcium Sparks // Science. 1995. Vol. 270, № 5236. P. 633–637.
- Knot H.J., Nelson M.T. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure // J. Physiol., 1998. Vol. 508, № 1. P. 199–209.
- Filosa J.A. et al. Local potassium signaling couples neuronal activity to vasodilation in the brain // Nat. Neurosci. 2006. Vol. 9, № 11. P. 1397–1403.
- Jackson W.F. Potassium channels in the peripheral microcirculation // Microcirculation. 2005. Vol. 12, № 1. P. 113–127.
- Bradley K.K. et al. Kir2.1 encodes the inward rectifier potassium channel in rat arterial smooth muscle cells // J. Physiol. 1999. Vol. 515 ( Pt 3, № 1999. P. 639–651.
- Quayle J.M., Dart C., Standen N.B. The properties and distribution of inward rectifier potassium currents in pig coronary arterial smooth muscle // J. Physiol. 1996. Vol. 494, № 3. P. 715–726.
- Longden T.A. et al. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow // Nat. Neurosci. 2017. Vol. 20, № 5. P. 717–726.
- Kim S.G., Ogawa S. Biophysical and physiological origins of blood oxygenation level-dependent fMRI signals // J. Cereb. Blood Flow Metab. 2012. Vol. 32, № 7. P. 1188–1206.
- Petzold G.C., Murthy V.N. Role of astrocytes in neurovascular coupling // Neuron. Elsevier Inc., 2011. Vol. 71, № 5. P. 782–797.
- Sonkusare S.K. et al. Inward rectifier potassium (Kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators // J. Physiol. 2016. Vol. 594, № 12. P. 3271–3285.
- Smith P.D. et al. K<subIR channels function as electrical amplifiers in rat vascular smooth muscle // J. Physiol. 2008. Vol. 586, № 4. P. 1147–1160.
- Kwan H.-Y. et al. Depletion of intracellular Ca2+ stores sensitizes the flow-induced Ca2+ influx in rat endothelial cells // Circ. Res. 2003. Vol. 92, № 3. P. 286–292.
- Ahn S.J. et al. Inwardly rectifying K+ channels are major contributors to flow-induced vasodilatation in resistance arteries // J. Physiol. 2017. Vol. 595, № 7. P. 2339–2364.
- Looft-Wilson R.C. et al. Flow does not alter eNOS phosphoryation at Ser1179 or Thr495 in preconstricted mouse mesenteric arteries // Physiol. Rep. 2018. Vol. 6, № 17. P. 1–11.
- Tristani-Firouzi M., Etheridge S.P. Kir 2.1 channelopathies: The Andersen-Tawil syndrome // Pfl?gers Arch. - Eur. J. Physiol. 2010. Vol. 460, № 2. P. 289–294.
- Dahal G.R. et al. An inwardly rectifying K+ channel is required for patterning // Development. 2012. Vol. 139, № 19. P. 3653–3664.
- Belus M.T. et al. Kir2.1 is important for efficient BMP signaling in mammalian face development // Dev. Biol. Elsevier Inc., 2018. Vol. S0012-1606, № 17. P. 30829–1.
- Wilde A.A.M., Bezzina C.R. Genetics of cardiac arrhythmias // Heart. 2005. Vol. 91, № 10. P. 1352–1358.
- Priori S.G. et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene // Circ. Res. 2005. Vol. 96, № 7. P. 800–807.
- Deo M. et al. KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia // Proc. Natl. Acad. Sci. 2013. Vol. 110, № 11. P. 4291–4296.
- Patel C., Yan G.X., Antzelevitch C. Short QT syndrome: From bench to bedside // Circ. Arrhythmia Electrophysiol. 2010. Vol. 3, № 4. P. 401–408.
- Cheung C.-L. et al. Genome-wide association study identifies a susceptibility locus for thyrotoxic periodic paralysis at 17q24.3 // Nat. Genet. 2012. Vol. 44, № 9. P. 1026–1029.
- Jongjaroenprasert W. et al. A genome-wide association study identifies novel susceptibility genetic variation for thyrotoxic hypokalemic periodic paralysis // J. Hum. Genet. 2012. Vol. 57, № 5. P. 301–304.
- Hille B. Ion channels of excitable membranes // Sunderland, Mass: Sinauer Associates, Inc. p. 5. ISBN 0-87893-321-2. Third edit. Sinauer Associates, Inc., 1992. Vol. Sunderland, № Third Edition. 814 p.
- Hille B., Schwarz W. Potassium channels as multi-ion single-file pores // J. Gen. Physiol. 1978. Vol. 72, № 4. P. 409–442.
- Fang Y. et al. Functional expression of Kir2.x in human aortic endothelial cells: the dominant role of Kir2.2 // Am. J. Physiol. Physiol. 2005. Vol. 289, № 5. P. C1134–C1144.
Телеграм: t.me/ainewsline
Источник: medach.pro