Старение — плата за подавление раковых опухолей?
МЕНЮ
Главная страница
Поиск
Регистрация на сайте
Помощь проекту
Архив новостей
ТЕМЫ
Новости ИИ
Городские сумасшедшие
ИИ в медицине
ИИ проекты
Искусственные нейросети
Искусственный интеллект
Слежка за людьми
Угроза ИИ
ИИ теория
Компьютерные науки
Машинное обуч. (Ошибки)
Машинное обучение
Машинный перевод
Нейронные сети начинающим
Психология ИИ
Реализация ИИ
Реализация нейросетей
Создание беспилотных авто
Трезво про ИИ
Философия ИИ
Генетические алгоритмы
Капсульные нейросети
Основы нейронных сетей
Промпты. Генеративные запросы
Распознавание лиц
Распознавание образов
Распознавание речи
Творчество ИИ
Техническое зрение
Чат-боты
Авторизация
2018-01-13 10:00
В нашем организме существуют обновляющиеся ткани, в которых есть пул постоянно делящихся клеток, которые заменяют отработанные или погибающие клетки. Такие клетки есть в криптах кишечника, в базальном слое эпителия кожи, в костном мозге (кроветворные клетки). Обновление клеток может происходить довольно интенсивно: так, клетки соединительной ткани в поджелудочной железе заменяются каждые 24 часа, клетки слизистой желудка — каждые три дня, лейкоциты — каждые 10 дней, клетки кожи — каждые шесть недель, примерно 70 г пролиферирующих клеток тонкого кишечника удаляется из организма ежедневно [1].
Стволовые клетки, существующие практически во всех органах и тканях, способны делиться неограниченно. Регенерация тканей происходит за счет пролиферации стволовых клеток, которые могут не только делиться, но и дифференцироваться в клетки той ткани, регенерация которой происходит. Стволовые клетки есть в миокарде, в головном мозге (в гипокампе и в обонятельных луковицах) и в других тканях. Это открывает большие надежды в плане лечения нейродегенеративных заболеваний и инфаркта миокарда
Постоянно обновляющиеся ткани способствуют увеличению продолжительности жизни. При делении клеток происходит омоложение тканей: новые клетки приходят на место поврежденных, при этом интенсивнее происходит репарация (устранение повреждений ДНК) и возможна регенерация при повреждении тканей. Не удивительно, что у позвоночных значительно выше продолжительность жизни, чем у беспозвоночных — тех же насекомых, у которых во взрослом состоянии клетки не делятся.
Но в то же время обновляющиеся ткани подвержены гиперпролиферации, что ведет к образованию опухолей, в том числе — злокачественных. Это происходит из-за нарушений регуляции деления клеток и повышенной частоты мутагенеза в активно делящихся клетках. По современным представлениям, чтобы клетка приобрела свойство злокачественности, ей необходимо
* — Стоит, в прочем, помнить, что мутация мутации рознь, и согласно новейшим геномным исследованиям в каждом поколении человек приобретает около 60 новых мутаций (которых не было в ДНК у его родителей). Очевидно, что большая часть из них вполне нейтральная (см. «Перевалило за тысячу: третья фаза геномики человека»). — Ред.
В целях защиты от самого себя, в организме сформировались специальные клеточные механизмы супрессии опухолей. Один из них — репликативное старение клеток (сенесценция), заключающееся в необратимой остановке деления клетки в стадии G1 клеточного цикла. При старении клетка перестает делиться: она не реагирует на ростовые факторы и становится устойчивой к апотозу.
Лимит Хейфлика
Феномен старения клеток был впервые открыт в 1961 г. Леонардом Хейфликом с коллегами на культуре фибробластов. Оказалось, что клетки в культуре фибробластов человека при хороших условиях живут ограниченное время и способны удваиваться примерно 50±10 раз, — и это число стали называть лимитом Хейфлика [6, 7]. До открытия Хейфлика господствовала точка зрения, что клетки бессмертны, а старение и смерть — это свойство организма в целом.
Эта концепция считалась неопровержимой во многом благодаря экспериментам Карреля, который поддерживал культуру клеток сердца цыпленка 34 года (ее выбросили лишь после его смерти). Однако, как выяснилось впоследствии, бессмертие культуры Карреля было артефактом, поскольку вместе с эмбриональной сывороткой, которая добавлялась в культуральную среду для роста клеток, туда попадали и сами эмбриональные клетки (и, скорее всего, культура Карреля стала уже далеко не тем, чем была в начале).
По-настоящему бессмертными являются раковые клетки. Так, клетки HeLa, выделенные в 1951 г. из опухоли шейки матки Генриетты Лакс*, до сих пор используются цитологами (в частности, c помощью клеток HeLa была разработана вакцина против полиомиелита). Эти клетки даже побывали в космосе.
* — О захватывающей истории бессмертия Генриетты Лакс см. в статье «Бессмертные клетки Генриетты Лакс», а также «Наследники клеток HeLa». — Ред.
Как выяснилось, лимит Хейфлика зависит от возраста: чем старше человек, тем меньшее число раз удваиваются его клетки в культуре. Интересно, что замороженные клетки при разморозке и последующем культивировании как будто помнят число делений до замораживания. Фактически, внутри клетки существует «счетчик делений», и по достижении определенного предела (лимита Хейфлика) клетка перестает делиться — становится сенесцентной. Сенесцентные (старые) клетки имеют специфическую морфологию — они крупные, уплощенные, с большими ядрами, сильно вакуолизированы, у них меняется профиль экспрессии генов. В большинстве случаев они устойчивы к апоптозу.
Однако старение организма нельзя свести только к старению клеток. Это значительно более сложный процесс. Старые клетки есть и в молодом организме, но их мало! Когда же с возрастом сенесцентные клетки накапливаются в тканях, начинаются дегенеративные процессы, которые приводят к возраст-зависимым заболеваниям. Один из факторов этих заболеваний — так называемое старческое «стерильное» воспаление, которое связано с экспрессией провоспалительных цитокинов старыми клетками.
Еще один важный фактор биологического старения — строение хромосом и их кончиков — теломеров.
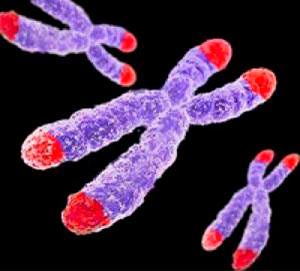
Рисунок 1. Теломеры — концевые участки хромосом. Поскольку хромосом у человека 23 пары (то есть, 46 штук), теломер получается 92.
Теломерная теория старения
В 1971 году наш соотечественник Алексей Матвеевич Оловников предположил, что лимит Хейфлика связан с «недорепликацией» концевых участков линейных хромосом (они имеют специальное название — теломеры). Дело в том, что в каждом цикле деления клетки теломеры укорачиваются из-за неспособности ДНК-полимеразы синтезировать копию ДНК с самого кончика [8, 9]. Кроме того, Оловников предсказал существование теломеразы (фермента, добавляющего повторяющиеся последовательности ДНК на концы хромосом), исходя из того факта, что иначе в активно делящихся клетках ДНК быстро бы «съелась», и генетический материал был бы утерян. (Проблема в том, что активность теломеразы угасает в большинстве дифференцированных клеток.)
Теломеры (рис. 1) играют важную роль: они стабилизируют кончики хромосом, которые иначе, как говорят цитогенетики, стали бы «липкими», т.е. подверженными разнообразным хромосомным аберрациям, что приводит к деградации генетического материала. Теломеры состоят из повторяющихся
Незащищенные концы хромосом воспринимаются клеткой как повреждение генетического материала, что активирует репарацию ДНК. Теломерный комплекс вместе с шелтерином «стабилизирует» хромосомные кончики, защищая всю хромосому от разрушения. В сенесцентных клетках критическое укорочение теломер нарушает эту защитную функцию [12, 13], в связи с чем начинают формироваться хромосомные аберрации, которые часто приводят к малигнизации. Чтобы этого не произошло, специальные молекулярные механизмы блокируют клеточное деление, и клетка переходит в состояние сенесцентности — необратимой остановки клеточного цикла. При этом клетка гарантированно не может размножаться, а значит, не сможет и сформировать опухоль. В клетках с нарушенной способностью к сенесценции (которые размножаются, несмотря на дисфункцию теломер), образуются хромосомные аберрации.
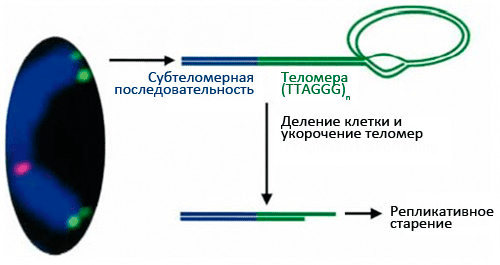
Рисунок 2. Состав и структура теломер. Многократное деление клетки в случае отсутствия активности теломеразы ведет к укорочению теломер и репликативному старению.
Длина теломер и скорость их укорочения зависит от возраста. У человека длина теломер варьирует от 15 тысяч нуклеотидных пар (т.н.п.) при рождении до 5 т.н.п. при хронических заболеваниях. Длина теломер максимальна у
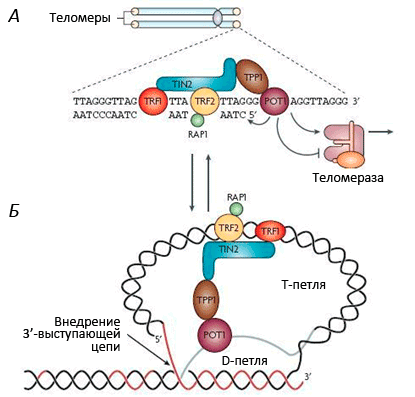
Рисунок 3. Строение теломерного комплекса (шелтерина). Теломеры находятся на концах хромосом и состоят из тандемных повторов TTAGGG, которые заканчиваются
Теломеры укорачиваются у разных людей с разной скоростью. Так, на эту скорость сильно влияют стрессы. Э. Блекберн (лауреат Нобелевской премии по физиологии и медицине 2009 г.) установлено, что женщины, постоянно испытывающие стресс (например, матери хронически больных детей), имеют значительно более короткие теломеры по сравнению со сверстницами (примерно на десять лет!). Лабораторией Э. Блекберн разработан коммерческий тест для определения «биологического возраста» людей на основании длины теломер.
Любопытно, что у мышей очень длинные теломеры
При сравнении длины теломер и теломеразной активности у разных млекопитающих оказалось, что виды, для которых характерно репликативное старение клеток, имеют большую продолжительность жизни и большой вес. Это, например, киты, продолжительность жизни которых может достигать 200 лет. Таким организмам репликативное старение просто необходимо, поскольку слишком большое число делений порождает множество мутаций, с которыми необходимо как-то бороться. Предположительно, репликативное старение и есть такой механизм борьбы, который сопровождается к тому же репрессией теломеразы [16].
Старение диференцированных клеток происходит иначе. Стареют и нейроны, и кардиомиоциты, а ведь они не делятся! Например, в них накапливается липофусцин — старческий пигмент, который нарушает функционирование клеток и запускает апоптоз. В клетках печени и селезенки с возрастом накапливается жир.
Связь репликативного старения клеток со старением организма, строго говоря, не доказана, но возрастная патология сопровождается и старением клеток (рис. 4). Злокачественные новообразования пожилого возраста в большинстве своем связаны с обновляемыми тканями. Онкологические заболевания в развитых странах — одна из основных причин заболеваемости и смертности, причем независимым фактором риска раковых заболеваний является просто... возраст. Число смертей от опухолевых заболеваний увеличивается с возрастом по экспоненте, так же как и общая смертность. Это говорит нам, что между старением и канцерогенезом существует фундаментальная связь.
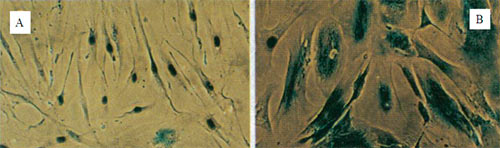
Рисунок 4. Гистохимически окрашенные на наличие ?-галактозидазной активности фибробласты человека линии WI-38. A: молодые; B: старые (сенесцентные).
Теломераза — фермент, который был предсказан
В организме должен существовать механизм, компенсирующий укорочение теломер, — такое предположение сделал А.М. Оловников. Действительно, в 1984 г. такой фермент был открыт Кэрол Грейдер и назван теломеразой. Теломераза (рис. 5) — это обратная транскриптаза, которая увеличивает длину теломер, компенсируя их недорепликацию. В 2009 году Э. Блэкберн, К. Грэйдер и Д. Шостак за открытие этого фермента и цикл работ по изучению теломер и теломеразы была присуждена Нобелевская премия (см: «„Нестареющая“ Нобелевская премия: в 2009 году отмечены работы по теломерам и теломеразе»).
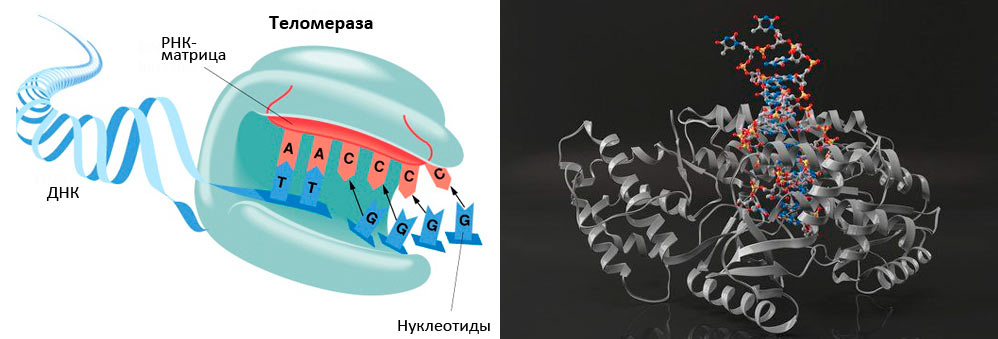
Рисунок 5. Теломераза содержит каталитический компонент (обратную транскриптазу ТERT), теломеразную РНК (hTR или TERC), содержащую две копии теломерного повтора и являющуюся матрицей для синтеза теломеров, и белок дискерин.
По данным Э. Блекберн, теломераза участвует в регуляции активности примерно 70 генов. Теломераза активна в зародышевых и эмбриональных тканях, в стволовых и пролиферирующих клетках. Ее обнаруживают в 90% раковых опухолей, что обеспечивает неудержимое размножение раковых клеток. В настоящее время среди препаратов, которые используют для лечения рака, есть и ингибитор теломеразы. Но в большинстве соматических клеток взрослого организма теломераза не активна.
В состояние сенесценции клетку могут привести многие стимулы — дисфункция теломер, повреждения ДНК, причиной которых могут быть мутагенные воздействия окружающей среды, эндогенные процессы, сильные митогенные сигналы (сверхэкспрессия онкогенов Ras, Raf, Mek, Mos, E2F-1 и др.), нарушения хроматина, стрессы и др. Фактически, клетки перестают делиться — становятся сенесцентными — в ответ на потенциально вызывающие рак события.
Страж генома
Дисфункция теломер, которая происходит при их укорачивании либо нарушении работы шелтерина, активирует белок р53. Этот транскрипционный фактор приводит клетку в состояние сенесценции, либо вызывает апоптоз. При отсутствии р53 развивается нестабильность хромосом, характерная для карцином человека. Мутации в белке р53 обнаруживаются в 50% аденокарцином груди и в
Теломераза реактивируется в большинстве опухолей эпителиального происхождения, которые характерны для пожилых людей. Считается, что реактивация теломеразы — важный этап злокачественных процессов, поскольку это позволяет раковым клеткам «не обращать внимания» на лимит Хейфлика. Дисфункция теломер способствует хромосомным слияниям и аберрациям, что в отсутствии p53 чаще всего приводит к злокачественным новообразованиям.
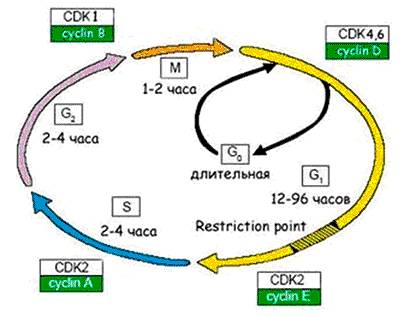
Рисунок 6. Схема клеточного цикла. Клеточный цикл подразделяют на четыре стадии: 1. G1 (предсинтетическая) — период, когда клетка готовится к репликации ДНК. В этой стадии может произойти остановка клеточного цикла в случае обнаружения повреждений ДНК (на время репарации). Если обнаруживаются ошибки в репликации ДНК, и они не могут быть исправлены репарацией, клетка не переходит на стадию S. 2. S (cинтетическая) — когда происходит репликация ДНК. 3. G2 (постсинтетическая) — подготовка клетки к митозу, когда происходит проверка точности репликации ДНК; если обнаружены недореплицированные фрагменты или другие нарушения в синтезе, переход на следующую стадию (митоз) не происходит. 4. М (митоз) — формирование клеточного веретена, сегрегация (расхождение хромосом) и формирование двух дочерних клеток (собственно деление).
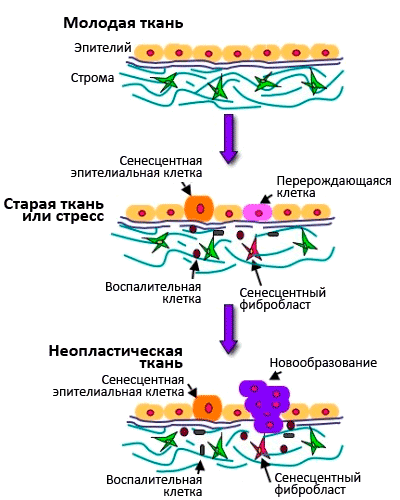
Рисунок 7. Взаимосвязь между старением клеток и старением организма.
О молекулярных механизмах старения клеток
Чтобы были понятны молекулярные механизмы перехода клетки в состояние сенесцентности, я напомню вам, как происходит деление клетки.
Процесс размножения клеток называют пролиферацией. Время существования клетки от деления до деления именуют клеточным циклом. Процесс пролиферации регулируется как самой клеткой — аутокринными ростовыми факторами, — так и ее микроокружением — паракринными сигналами.
Активация пролиферации происходит через клеточную мембрану, в которой присутствуют рецепторы, воспринимающие митогенные сигналы — это в основном ростовые факторы и межклеточные контактные сигналы. Ростовые факторы обычно имеют пептидную природу (к настоящему времени их известно около 100). Это, например, фактор роста тромбоцитов, который участвует в тромбообразовании и заживлении ран, эпителиальный фактор роста, различные цитокины — интерлейкины, фактор некроза опухолей, колониестимулирующие факторы и т.д. После активации пролиферации клетка выходит из фазы покоя G0 и начинается клеточный цикл [19] (см. рис. 6).
Клеточный цикл регулируется циклин-зависимыми киназами, разными для каждой стадии клеточного цикла. Они активируются циклинами и инактивируются рядом ингибиторов. Цель такой сложной регуляции — обеспечить синтез ДНК с как можно меньшим числом ошибок, чтобы и дочерние клетки имели абсолютно идентичный наследственный материал. Проверка правильности копирования ДНК осуществляется в четырех «контрольных точках» цикла: если обнаруживаются ошибки, то клеточный цикл останавливается, и включается репарация ДНК. Если нарушения структуры ДНК удается исправить — клеточный цикл продолжается. Если нет — клетке лучше «покончить с собой» (путем апоптоза), чтобы избежать вероятности превращения в раковую.
Молекулярные механизмы, приводящие к необратимой остановке клеточного цикла, контролируются генами-супрессорами опухолей, среди которых p53 и pRB, связанные с ингибиторами циклин-зависимых киназ. Супрессию клеточного цикла в фазе G1 осуществляет белок p53, действующий через ингибитор циклин-зависимой киназы р21. Транскрипционный фактор р53 активируется при повреждениях ДНК, и функция его заключается в удалении из пула реплицирующихся клеток тех, которые являются потенциально онкогенными (отсюда и прозвище р53 — «страж генома»). Данное представление подтверждается тем фактом, что мутации р53 обнаруживают в ~50% случаев злокачественных опохолей. Другое проявление активности р53 связано с апоптозом наиболее поврежденных клеток.
Сенесценция клеток и возраст-зависимые заболевания
Сенесцентные клетки накапливаются с возрастом и способствуют возрастным заболеваниям. Они снижают пролиферативный потенциал ткани и истощают пул стволовых клеток, что приводит к дегенеративным нарушениям ткани и снижает способность к регенерации и обновлению.
Сенесцентные клетки характеризуются специфической экспрессией генов: они секретируют воспалительные цитокины и металлопротеиназы, разрушающие межклеточный матрикс. Получается, что старые клетки обеспечивают вялотекущее старческое воспаление, а накопление старых фибробластов в коже служит причиной возрастного снижения способности к заживлению ран (см. рис. 7). Старые клетки также стимулируют пролиферацию и малигнизацию близлежащих предраковых клеток, благодаря секреции эпителиального фактора роста [20].
Сенесцентные клетки накапливаются во многих тканях человека, присутствуют в атеросклеротических бляшках, в язвах кожи, в пораженных артритом суставах, а также в доброкачественных и пренеопластических гиперпролиферативных поражениях простаты и печени. При облучении раковых опухолей некоторые клетки также переходят в состояние сенесценции, тем самым обеспечивая рецидивы заболевания.
Таким образом, клеточное старение демонстрирует эффект отрицательной плейотропии, суть которого состоит в том, что хорошее для молодого организма, может стать плохим для старого. Самый яркий пример — процессы воспаления. Выраженная реакция воспаления способствует быстрому выздоровлению молодого организма при инфекционных заболеваниях. В пожилом же возрасте активные воспалительные процессы приводят к возрастным заболеваниям. Сейчас принято считать, что воспаление играет определяющую роль практически при всех возраст-зависимых заболеваниях, начиная с нейродегенеративных.
Получается парадокс: старение клеток в молодом организме предохраняет от рака, а в старом — способствует ему! В настоящее время в США в клинике Майо ведутся исследования влияния «ликвидации» старых клеток из организма. На животных получены обнадеживающие результаты об увеличении продолжительности жизни и замедлении клинических проявлений возраст-зависимых заболеваний, если из тканей старых животных селективно устранять сенесцентные клетки — хороших граждан, но плохих соседей.
Литература
- Macieira-Coelho A. (2011). Cell division and ageing of the organism. Biogerontology 12,
503–515; ; - Была клетка простая, стала стволовая;
- Ствол и ветки: стволовые клетки;
- Нобелевская премия по физиологии и медицине (2012): индуцированные стволовые клетки;
- Егоров Е.Е. Роль теломер и теломеразы в процессах клеточного старения и канцерогенеза. // Дисс. д-ра биол. наук, М., 2003. 300 с.;
- Hayflick L., Moorhead P.S. (1961). The serial cultivation of human diploid cell strain. Exp. Cell Res. 25,
585–621; ; - Shay J.W., Wright W.E. (2000). Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell. Biol. 1,
72–76; ; - Оловников А.М. (1971). Принцип маргинотомии в матричном синтезе полинуклеотидов. Докл. акад. наук СССР 201,
1496–1499; ; - Olovnikov A.M. (1973). A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 41,
181–190; ; - Greider C.W. (1994). Mammalian telomere dynamics: healing, fragmentation shortening and stabilization. Curr. Opin. Genet. Dev. 4,
203–211; ; - Коряков Д.Е., Жимулев И.Ф. (2009). Хромосомы. Структура и функции. СО РАН, 256 с;
- de Lange T. (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19,
2100–2110; ; - Deng Y., Chang S. (2007). Role of telomeres and telomerase in genomic instability, senescence and cancer. Lab. Invest. 87,
1071–1076; ; - http://biomolecula.ru#;
- Анисимов В.Н. (2008). Молекулярные и физиологические механизмы старения. Сп.-Б.: «Наука», том 1 стр. 465, том 2, стр. 473;
- Gomes N.M.V., Shay J.W., Wright W.E. (2010). Telomeres and Telomerase. The Comparative Biology of Aging (ed. Wolf N.S.). Springer, p.
227–259; ; - Shay J.W., Wright W.E. (2004). Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis 26,
867–874; ; - Shay J.W., Wright W.E. (2001). Forward: aging and cancer: are telomeres and telomerase the connection? In: Telomerase, Aging and Disease (ed. Mattson M.P.). Baltimore, MD: Elsevier, p. 231;
- Фаллер В.М.., Шилдс Д. (2006). Молекулярная биология клетки. М.: «Бином-Пресс», 235 стр.;
- Campisi J. (2005). Senescent cells, tumor suppression, and organismal aging: good citizens, bad Neighbors. Cell 120,
513–522; ; - Suzuki M., Boothman D.A. (2008). Stress-induced premature senescence (influence of SIPS on radiotherapy). J. Rad. Res. 49,
105–112. .
Телеграм: t.me/ainewsline
Источник: biomolecula.ru