Энергия шванновских клеток «в помощь» погибающим нейронам
МЕНЮ
Искусственный интеллект
Поиск
Регистрация на сайте
Помощь проекту
ТЕМЫ
Новости ИИ
Голосовой помощник
Городские сумасшедшие
ИИ в медицине
ИИ проекты
Искусственные нейросети
Слежка за людьми
Угроза ИИ
Компьютерные науки
Машинное обуч. (Ошибки)
Машинное обучение
Машинный перевод
Нейронные сети начинающим
Реализация ИИ
Реализация нейросетей
Создание беспилотных авто
Трезво про ИИ
Философия ИИ
Генетические алгоритмы
Капсульные нейросети
Основы нейронных сетей
Распознавание лиц
Распознавание образов
Распознавание речи
Техническое зрение
Чат-боты
Авторизация
2020-12-09 13:10

В новом исследовании показано, что сразу после повреждения аксона в шванновских клетках усиливаются процессы гликолиза для обеспечения аксонов энергией и предотвращения их дегенерации. Авторы также определяют возможные цели терапевтического вмешательства, на которые можно воздействовать для обеспечения защиты аксонов.
Повреждение аксона — инициальное событие при некоторых острых и хронических нейродегенеративных заболеваниях, что часто ведет к гибели нейронов [1]. Таким образом, сохранение целостности аксонов имеет решающее значение для предотвращения быстрого развития этого необратимого процесса. В то время как центральная роль нейронов в процессе дегенерации была тщательно исследована [1, 2], недавние работы показывают, что шванновские клетки, миелинизирующие глиоциты периферической нервной системы (ПНС), также играют важную роль в правильной активации регенерационных процессов в нервной ткани [3]. Тем не менее, доподлинно неизвестно, как и непосредственно ли сами клетки Шванна и аксоны нейронов влияют друг на друга, способствуя регенерации (как это происходит в ЦНС) [4, 5]. Эти вопросы поднимаются в новой работе Babetto с соавт. в текущем выпуске «Nature Neuroscience».
После травмы нейроны и глиальные клетки участвуют в сложной программе внутриклеточных событий по контролю аксональной дегенерации, в конечном итоге приводящей к немедленной активации программы регенерации, направленной на предотвращение гибели нейронов [2]. Для функционирования этой программы необходима активация нескольких молекулярных процессов, организованных в сложную сигнальную систему, ключевым среди которых является выделение кальция из внутриклеточного нейронального пространства [7]. В свою очередь, клетки Шванна теряют черты дифференцировки (процесс т. н. дедифференцировки), в них активируется специальная программа аутофагии, и они мигрируют вдоль растущей сосудистой оболочки, образуя полосы Бюнгнера, которые в конечном итоге служат направлением для роста поврежденных аксонов [3]. Если в конце процесса аксоны успешно достигают своей цели, клетки Шванна вновь дифференцируются и ремиелинизируют аксоны. На молекулярном уровне в глиоцитах было выявлено несколько факторов, среди которых транскрипционный фактор c-Jun, играющий ведущую роль на протяжении реализации всей программы глиальной репарации [8].
Помимо выделения кальция, в аксонах активируется локальная трансляция мРНК для регуляции обратной сигнализации (от аксона к телу нейрона) и модуляции ответа на повреждение нейрона. Последний процесс крайне энергоемкий, одну из ролей в его контроле играет комплекс 1 мишени рапамицина в клетках млекопитающих (mTORC1) [9]. Для поддержания процесса восстановления нейроны должны перейти в особый регенерационный режим и активировать серию транскрипционных событий, способствующих экспрессии определенного набора генов [2]. Следует отметить, что эпигенетические модификации также являются крайне важными для описанных изменений и для поддержки функции конусов роста [2].
Все эти процессы требуют большого количества энергии. Фенотип шванновских клеток превращается в регенераторный, в то время как нейроны переходят из катаболического состояния в анаболическое для обеспечения синтеза всех макромолекул, необходимых для развития регенерирующих аксонов [2, 3].
В своем новом исследовании [6] Babetto с соавт. показывают, что вскоре после аксотомии (иссечение аксона целиком или его части — прим. перев.), когда в шванновских клетках активируется программа дедифференцировки для формирования ответной реакции на немедленные изменения, происходящие вслед за повреждением [3], сами шванновские клетки также адаптируют свою метаболическую активность под возросшую потребность в энергии. И действительно, резко повышается активность ферментов гликолиза для синтеза пирувата и лактата, необходимых в качестве энергетической поддержки аксонов для предотвращения их дегенерации (рис. 1).
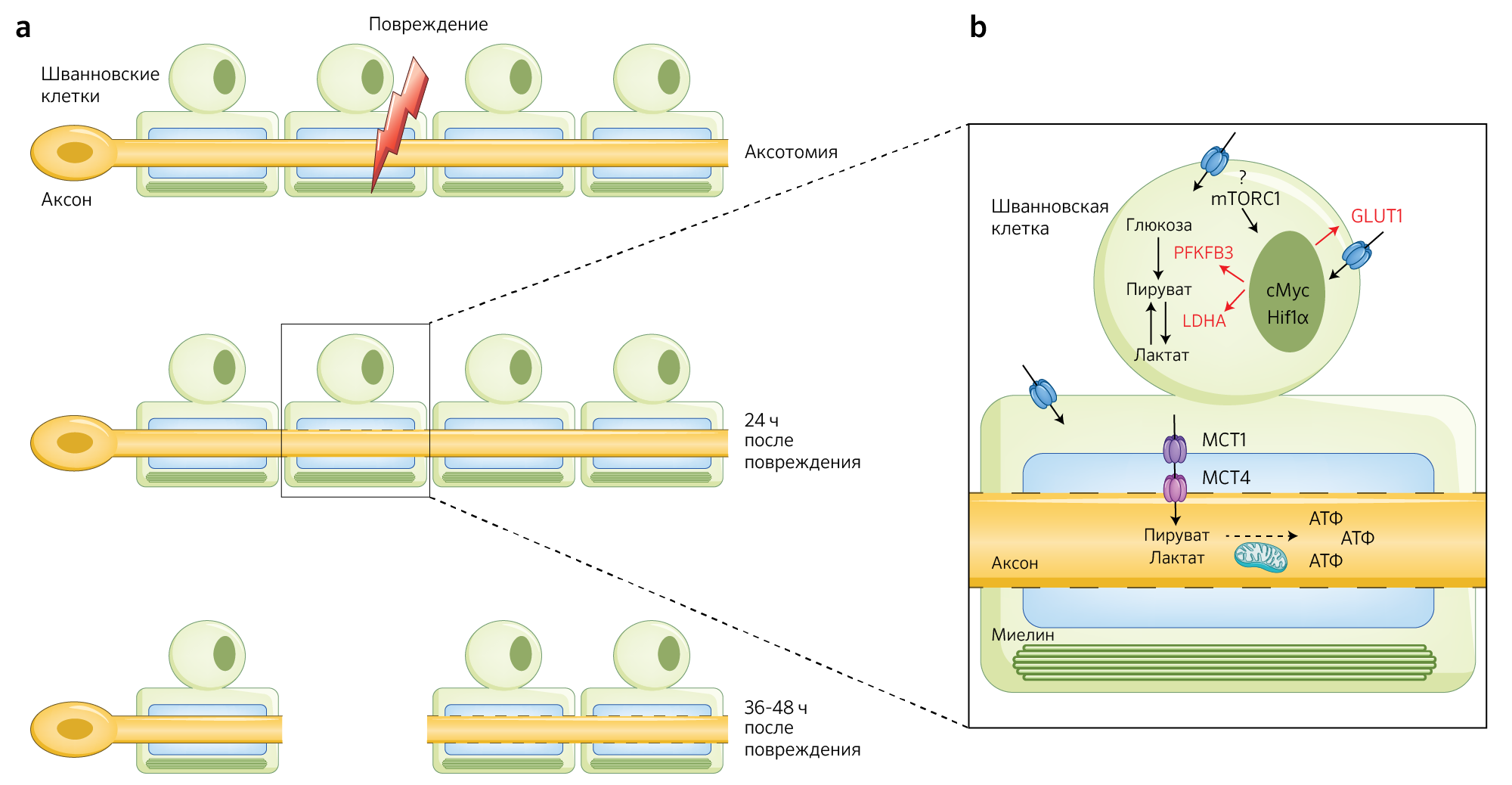
Рис.1 | Переключение на гликолитический путь окисления в шванновских клетках при Валлеровой дегенерации
а — Схематичное изображение первых стадий аксональной дегенерации после повреждения. SC — шванновские клетки.
b — Сразу после травмы в клетках Шванна наблюдается резкое повышение гликолиза под контролем сигнального каскада — оси mTORC1–Hif1?–c-Myc.
Преобладание гликолиза приводит к избыточному синтезу пирувата и лактата, которые транспортируются в аксоны для поддержания их функциональной целостности и, вероятно, служат для поддержания активного процесса регенерации.
Используя модель травматического повреждения аксонов in vitro, авторы показали, что простого присутствия клеток Шванна достаточно для замедления аксональной дегенерации, что, в таком случае, ведет к лишь частичной фрагментации аксонов узла заднего корешка (УЗК) спинного мозга. Эксперименты, целью которых было определение механизма, лежащего в основе этого явления, показали, что клетки Шванна подвергаются метаболическому переключению. Оно характеризуется повышением экспрессии всех ферментов гликолиза. Примечательно, что повышение гликолитической активности в глиальных клетках в сочетании с временным повышением экспрессии ключевого ферментного комплекса-активатора гликолиза 6-фосфофрукто2-киназы/фруктоза-2,6-бифосфатазы 3 (PFKFB3) может служить источником энергии для нейронов, находящихся в стадии дегенерации.
Авторы нашли доказательства этой гипотезы in vivo. Им удалось наблюдать повышение поглощения глюкозы и потребления лактата в поврежденных седалищных нервах у мышей спустя почти 48 ч после аксотомии. Это сопровождалось повышенной экспрессией в шванновских клетках фермента GLUT1, ответственного за поглощение глюкозы, и лактатдегидрогеназы А (ЛДГА) — фермента, преобразующего пируват в лактат. Хотя метаболомный анализ выявил снижение промежуточных метаболитов гликолиза в поврежденных нервах (что предполагает быстрое потребление глюкозы), в клетках Шванна обнаружилось повышение экспрессии PDHK1 (киназа пируватдегидрогеназы 1 — фермент из семейства PDHK, фосфорилирующий и инактивирующий пируватдегидрогеназу). Данный факт свидетельствует о том, что в клетках Шванна пируват служит субстратом, служащим для накопления энергии (т. е. не происходит превращения его в ацетил-CoA для дальнейшего участия в аэробном пути окисления).
Чтобы подтвердить свои выводы, авторы провели нокаут экспрессии генов ключевых гликолитических ферментов в шванновских клетках. Вследствие этого у мышей-мутантов с удаленными генами Glut1, Pfkfb3 или LdhА в шванновских клетках (ответственных за миелинизацию аксонов) сразу же после аксотомии наблюдалась выраженная гибель аксонов нейронов. Эти данные являются убедительными доказательствами того, что метаболическое сообщение между клетками Шванна и аксонами служит для снабжения поврежденных нейронов пируватом и лактатом. Эти метаболиты необходимы для восстановления энергетического дефицита и для предотвращения (ограничения) аксональной дегенерации, аналогично роли олигодендроцитов в ЦНС [4, 5].
Интересно, что активация гена ErbB2 в изолированных клетках Шванна ведет к ускоренному накоплению лактата в супернатанте. Это привело авторов к постулату о том, что сигнальный путь «белок нейрегулин 1 — ген ErbB2» может находится вне зоны воздействия этого метаболического переключателя. Однако роль ErbB2 в Валлериановой дегенерации спорна [10, 11]. Таким образом, будущие исследования in vivo необходимы, чтобы выявить молекулярные механизмы, которые запускают описанные события, и определить, что вызывает метаболические сдвиги: дедифференцировка клеток Шванна или же изменения в глиально-аксональном взаимодействии.
Наблюдаемое накопление пирувата и лактата в клетках Шванна ставит вопрос о том, какой может быть судьба этих двух метаболитов. Анализ in vitro выявил сильную индукцию переносчиков монокарбоксилата MCT1 и MCT4 в глиальных клетках вскоре после аксотомии. Данная находка подразумевает, что транспортировка пирувата и лактата к аксонам увеличивается сразу же после травмы. Ключевую роль метаболитов шванновских клеток в поддержании функционирования и в предотвращении дегенерации аксонов удалось подтвердить в экспериментах in vitro. Восстановление поврежденных нейронов УЗК с помощью пирувата (который обладает свойством проникновения сквозь клеточную мембрану) оказалось достаточной мерой для замедления дегенерации аксонов, в то время как добавление специфических ингибиторов MCT уменьшало выживаемость аксонов.
Следующим шагом стала попытка авторов выяснить, какие сигнальные пути могут активировать гипергликолитический фенотип в клетках Шванна. С помощью поиска фосфорилированных молекул после повреждения удалось выявить сигнальные пути AMPK и mTOR. AMPK регулирует экспрессию GLUT1 и PFKFB3, а mTOR контролирует экспрессию нескольких генов, участвующих в метаболизме глюкозы. Эксперименты in vivo подтвердили важную роль пути mTOR в аксональной защите. Действительно, удаление гена mTOR (а не AMPK) в отдельных шванновских клетках снизило выживаемость аксонов после аксотомии и понизило экспрессию GLUT1, PFKFB3 и ЛДГА. Следует отметить, что у этих мышей-мутантов также снизилась экспрессия мишеней ферментов mTOR: индуцируемый гипоксией фактор 1? (Hif1?) и c-Myc12. Интересно, что одной из характеристик раковых клеток, у которых также наблюдаются процессы дедифференцировки, является аномальное повышение поглощения и потребления глюкозы [12], что часто приводит к активации пути mTOR. Таким образом, с точки зрения метаболизма, интригующе звучит то, что процесс дедифференцировки клеток Шванна напоминает аналогичный процесс в опухолевых клетках.
Для определения того, какой компонент сигнального пути mTOR задействован, авторы вывели мышей-мутантов, у которых в шванновских клетках были удалены гены, кодирующие белки Raptor или же Rictor, являющиеся субъединицами mTORC1 и mTORC2, соответственно. Только у мутантов с отсутствующим Raptor наблюдалась сниженная выживаемость аксонов после аксотомии. Кроме того, экспериментально увеличив активность mTORC1 за счет селективного удаления гена TSC2 (кодирует комплекс туберозного склероза 2 или туберин) во взрослых шванновских клетках был замедлен распад миелина и дегенерация аксонов после травмы. Следует отметить, что у мышей-мутантов с крайне сниженной экспрессией TSC1 в зрелых шванновских клетках наблюдаются дефективные процессы ремиелинизации на 12-й день после повреждения нерва [13]. Независимо от того, обусловлены ли эти различающиеся результаты различными экспериментальными моделями, или тем фактом, что TCS1 и TSC2 в поврежденных клетках Шванна имеют различные мишени, или же различными анализируемыми временными точками, необходимы дополнительные исследования. Конечно, основываясь на других исследованиях [6], mTOR позиционируется как центральный узел программ регенерации аксонов и глиоцитов [7, 14].
В своем последнем эксперименте авторы проверили, будут ли работать сделанные ими молекулярно-биологические выводы in vivo (использовалась мышиная модель хронической аксональной дегенерации). В нем выключение TCS2 у зрелых клеток Шванна привело к смягчению неврологического дефицита у мышей.
В совокупности эти выводы свидетельствуют о том, что комплексы mTORC1, Hif1a и c-Myc являются составными элементами молекулярного пути, контролирующего метаболическое переключение, происходящее в клетках Шванна сразу после травмы. Эти выводы также указывают на то, что упреждающую активацию этого сигнального пути можно использовать в качестве потенциального практического приложения для нивелирования неврологического дефицита. Однако несколько вопросов остаются нерешенными. Способны ли шванновские клетки поддерживать метаболизм аксонов и на более поздних стадиях Валлериановой дегенерации? Связаны ли эти изменения с процессом обратной дифференцировки, который в шванновских клетках характеризуется повышенной экспрессией белка c-Jun (продукт гена JUN), как сообщалось ранее [15]? Как клетки Шванна, отличающиеся своей способностью к дедифференцировке, воспринимают происходящую дегенерацию нервной ткани и что лежит в основе их быстрой реакции? Если mTOR действительно является центральным узлом регенерации в ПНС, может ли ограниченная экспрессия этого белкового комплекса в нейронах ЦНС [1] быть достаточным объяснением плохой регенеративной способности ЦНС? Ответив на некоторые или на все эти вопросы, появится возможность узнать больше о механизмах, контролирующих регенерацию нервной ткани, и об удивительном сообщении между аксонами и клетками глии во время регенерации.
Источник: medach.pro