Еще раз об апоптозе
МЕНЮ
Искусственный интеллект
Поиск
Регистрация на сайте
Помощь проекту
ТЕМЫ
Новости ИИ
Голосовой помощник
Городские сумасшедшие
ИИ в медицине
ИИ проекты
Искусственные нейросети
Слежка за людьми
Угроза ИИ
Компьютерные науки
Машинное обуч. (Ошибки)
Машинное обучение
Машинный перевод
Реализация ИИ
Реализация нейросетей
Создание беспилотных авто
Трезво про ИИ
Философия ИИ
Генетические алгоритмы
Капсульные нейросети
Основы нейронных сетей
Распознавание лиц
Распознавание образов
Распознавание речи
Техническое зрение
Чат-боты
Авторизация
2018-10-20 11:06

Каждый день в организме погибает большое количество клеток, а на смену им образуется равное количество новых: таково условие для поддержания клеточного гомеостаза. Удалить необходимо ненужные, старые и потенциально опасные. Одним из известных механизмов клеточной гибели является апоптоз, представляющий собой программируемый процесс.
Иначе можно сказать, что смерть клетки происходит согласно контролируемой «суицидальной» программе, регулируемой на генетическом уровне. Кроме того, апоптоз характеризуется «аккуратностью» (в отличие от некроза): мембрана погибшей клетки остается целой, и, следовательно, содержимое клетки не покидает ее границ, а воспалительная реакция не активируется. Этот механизм имеет важное значение для медицины, так как лежит в основе развития многих заболеваний.
Когда активируется апоптоз?
Физиологические ситуации:
- Разрушение клеток в процессе эмбриогенеза. Начальный этап развития организма сопровождается образованием избыточного клеточного материала, уничтожение которого происходит путем апоптоза в строго определённых местах и времени. Иначе говоря, гистогенез и органогенез тесно связаны с активацией апоптоза. Пример: удаление перепонок между зачатками пальцев.
- Инволюция гормонозависимых тканей после прекращения гормональной стимуляции. Пример: разрушение эндометрия во время менструального цикла, атрофия яичников в период менопаузы, постлактационное уменьшение молочной железы и атрофия простаты после кастрации.
- Ликвидация потенциально опасных лимфоцитов, которые могут реагировать на собственные ткани.
- Смерть клеток, которые уже послужили во благо организму. Например, гибель нейтрофилов при остром воспалительном ответе и лимфоцитов в конце иммунного ответа.
Патологические ситуации:
- Поврежденная ДНК. Радиоактивные и цитотоксические противоопухолевые препараты, а также гипоксия могут повредить ДНК либо напрямую, либо через производство свободных радикалов. Если система репарации не может справиться с повреждением, то клетка активирует внутренние механизмы, которые индуцируют апоптоз. Это является лучшей стратегией, так как существует риск злокачественного перерождения клеток.
- Накопление неправильно сложенных белков в эндоплазматическом ретикулуме. Это явление называется эндоплазматическим стрессом. Неправильно сложенные белки могут появится из-за мутаций в генах или при повреждениях свободными радикалами. Апоптоз, возникающий в результате таких накоплений, наблюдается, например, при нейродегенеративных заболеваниях (болезнь Альцгеймера, Паркинсона и др.).
- Гибель клеток при вирусных инфекциях. Потеря инфицированных клеток также обусловлена апоптозом, который может быть вызван вирусами (при аденовирусной и ВИЧ-инфекциях) или иммунной системой хозяина (при вирусном гепатите). За последнее отвечают цитотоксические Т-лимфоциты, которые убивают инфицированные клетки, устраняя резервуары инфекционного заболевания. Этот же механизм, опосредованный Т-клетками, наблюдается при гибели опухолевых клеток и при отторжении трансплантата.
Внутриклеточный протеолитический каскад
Активируют апоптоз внутриклеточные ферменты — каспазы (caspases). Они относятся к классу протеолитических ферментов (протеазы), так как расщепляют пептидные связи в белках. Буква «с» в «caspases» указывает на то, что в активном центре протеаз находится аминокислота цистеин, «asp» — на то, что расщепление последовательности аминокислот происходит после остатка аспарагиновой кислоты. Каспазы в клетке находятся в неактивной форме (в виде проферментов) и активируются только в процессе апоптоза.
Существует два класса каспаз: инициаторные (каспазы-2, -8, -9 и -10) и эффекторные (каспазы-3, -6 и -7). Первые отвечают за начало апоптоза, вторые же регулируют расщепление клеточных компонентов. Процесс развивается, как каскад, то есть состоит из нескольких ферментативных реакций. Субстратом на каждой стадии является белок, который в результате реакции превращается в активный фермент. Этот фермент в свою очередь использует другой белок в качестве субстрата, превращая его в активный фермент. И так повторяется несколько раз.
Каспазами разрушается множество белков, среди которых белки ядерной пластинки и белок-ингибитор активности эндонуклеазы. Расщепление последнего ведет к тому, что эндонуклеаза начинает разрезать ДНК. Разрушаются белки цитоскелета и клеточной адгезии, которые соединяют клетки друг с другом. Такой каспазный каскад необратим.
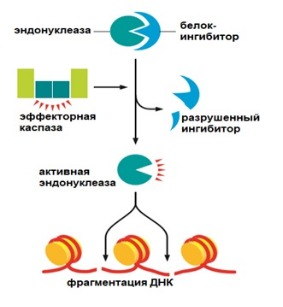
Рисунок 1 | Разрушение каспазой ингибитора эндонуклеазы и последствия. Апоптоз может протекать по двум различным путям — по внешнему и внутреннему (митохондриальному)
Внешний путь апоптоза
Этот путь запускается при связывании лиганда с рецептором смерти, находящимся на плазматической мембране различных клеток. Рецепторы смерти (death receptors — DR) бывают нескольких видов: TNF-R1, FAS (CD95), DR3, TRAIL-R1, TRAIL-R2 и др. Все они трансмембранные белки, содержащие внеклеточную часть — лиганд-связывающий домен — и внутриклеточную часть — домен смерти.
Иллюстрация такого пути — взаимодействие Fas рецептора на поверхности многих типов клеток с Fas-лигандом на цитотоксическом лимфоците. Домен смерти активированного рецептора объединяется с внутриклеточными белками FADD (Fas-associated death domain). Они в свою очередь объединяются с инициаторными каспазами, образуя сигнальный комплекс, вызывающий смерть (death-inducing signaling complex — DISC). Этот комплекс активирует инициаторные каспазы, которые затем включают в работу эффекторные каспазы, что дает начало апоптозу.
Существует ингибиторный белок, ограничивающий внешний путь. Этот белок называется FLIP. Он похож на инициаторную каспазу, но не обладает ее функцией. FLIP с каспазой-8 образует DISC, однако каспаза-8 не становится активной и апоптотический сигнал блокируется. Этот тормозный механизм помогает предотвратить нежелательную активацию внешнего пути.
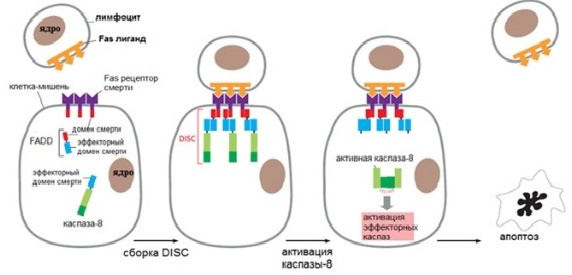
Рисунок 2 | Внешний путь апоптоза
Внутренний путь апоптоза зависит от митохондрий
Этот путь может быть запущен в ответ на повреждение ДНК, активацию онкогенов, избыток Ca2+ в клетке, отсутствие факторов роста (пептидный или стероидный гормон, стимулирующий рост и дифференцировку клетки), неправильно сложенные белки.
Активация пути ведет к повышению проницаемости наружной мембраны митохондрий. Из-за этого в цитоплазму выходят цитохром c и другие митохондриальные белки, которые инициируют апоптоз.
В норме они находятся в межмембранном пространстве этих органелл. Ключевой белок во внутреннем пути — цитохром с (компонент электрон-транспортной цепи). Выйдя в цитоплазму, он приобретает новые функции и присоединяется к фактору апоптотической протеазы 1 (apoptotic protease activating factor-1 — Apaf1).
Так образуется колесоподобная структура — апоптосома. Апоптосома активирует инициаторные каспазы-9, в свою очередь активирующие эффекторные каспазы, что дает начало апоптозу.
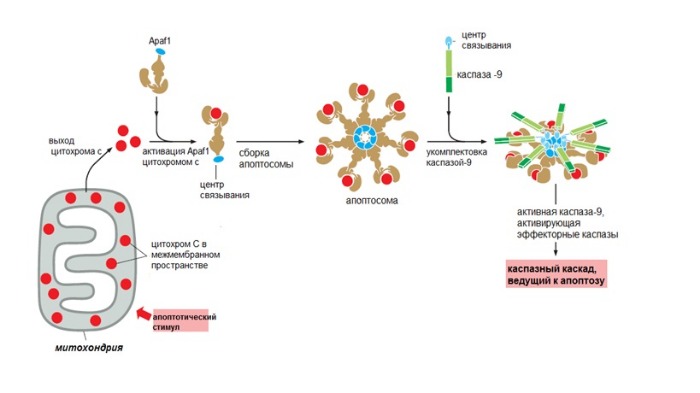
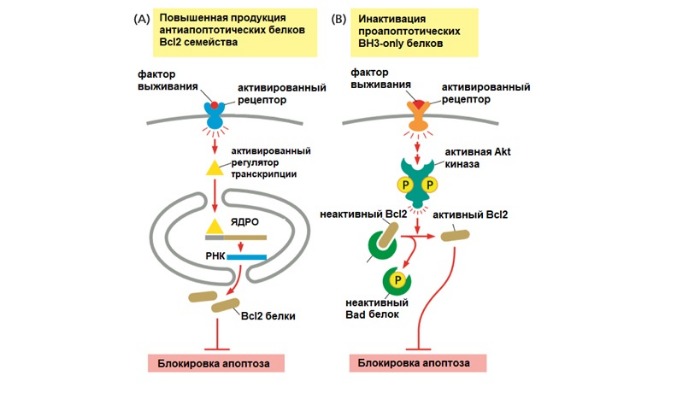
Апоптоз-регулируемый процесс
За внутренний путь апоптоза отвечают белки семейства Bcl2. Они контролируют выход проапоптотических белков из митохондрий (например, цитохром c). Название дано в честь гена белка Bcl2, который сверхэкспрессирован в некоторых лимфомах В-клеток (B cell lymphoma). В это семейство входят более 20 белков, которые могут быть разделены в три группы на основании их функций и количестве гомологичных доменов (Bcl2 Homology).
Первая группа — проапоптотические белки, которые увеличивают выход митохондриальных белков и запуск апоптоза.
Вторая группа — антиапоптотические белки, которые подавляют апоптоз, блокируя выход митохондриальных белков. Оба вида могут связываться друг с другом в различных комбинациях, подавляя свои функции. Баланс между активностью двух видов белков определяет, выживет ли клетка или погибнет по внутреннему пути апоптоза.
Антиапоптотическая группа представлена белками Bcl2 и BclXL, которые имеют четыре BH домена (BH1-4). Эти белки находятся на наружной мембране митохондрий и сохраняют ее непроницаемость. Таким образом это предотвращает утечку цитохрома c и других белков.
Проапоптотические белки — Bax и Bak. У них есть три BH домена (BH1-3). После своей активации Bax и Bak повышают проницаемость внешней мембраны митохондрий. Возможно, это происходит путем образования канала, что позволяет белкам выходить из межмембранного пространства в цитоплазму. Bak даже в отсуствие апоптотического сигнала связан с наружной мембраной митохондрий, а Bах локализован в цитозоле и транспортируется к митохондрии только после апоптотического сигнала.
Третья группа содержит (тоже проапототические) белки Bad, Bim, Bid, Puma и Noxa. Они имеют один BH домен (BH3), третий из четырех доменов BH, поэтому и получили название BH3 only proteins. Белки BH3-only играют ключевую роль в регулировании и стимулировании апоптоза и, таким образом, служат привлекательной целью терапевтического вмешательства. Следует отметить, что BH3 домен является единственным общим доменом для всех членов семейства Bcl2. Он опосредует взаимодействия между проапоптотическими и антиапоптотические белками.
Как происходит регуляция?
Факторы роста и другие сигналы выживания стимулируют выработку антиапоптотических белков. Они ингибируют апоптоз путем связывания проапоптотических белков на митохондриальной мембране. BH3-only белки, напротив, нейтрализуют активность антиапоптотических белков, таким образом способствуя собиранию проапоптотических белков Вах и Вак на поверхности митохондрии. Это приводит к выходу митохондриальных белков наружу.
Знаменитый белок р53 часто называют «стражем генома», потому что он в ответ на повреждение ДНК запускает апоптоз. Если повреждения не могут быть исправлены, белок р53 (опухолевый супрессор) накапливается в клетке и активирует транскрипцию генов, кодирующих BH3-only белки Puma и Noxa. Также p53 действует на митохондрии и взаимодействует с антиапоптотическим белком Bcl-xL.
Белок ВН3-only Bid связует оба пути апоптоза. В норме он неактивен. Но при активации внешнего пути каспаза-8 переводит белок Bid в активную форму. Bid перемещается к наружной мембране митохондрии и ингибирует антиапоптотические белки, тем самым увеличивая сигнал смерти.
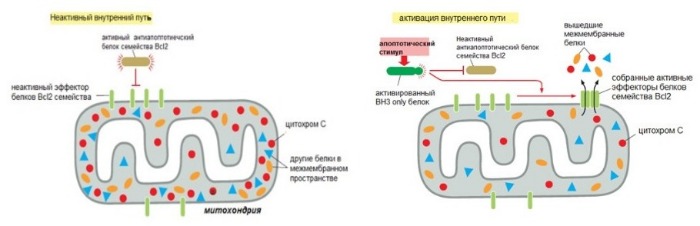
Рисунок 4 | Схема регуляции внутреннего пути апоптоза
Другие способы регуляции
Клетка использует надежные механизмы от ненужной активации каспаз. Например, защитником служит семейство белков-ингибиторов апоптоза (inhibitors of apoptosis — IAPs). У человека они представлены следующими видами: cIAP1 (BIRC2), cIAP2 (BIRC3), X-связанный IAP (XIAP) и др.
Одни из этих белков связывают и ингибируют активированные каспазы. Другие - помечают каспазы для разрушения протеосомами. Функция ингибиторов заключается в установлении порога, который каспазы должны преодолеть для активации апоптоза. Активность IAP может быть подавлена белками из межмембранного пространства митохондрий, такими как Omi/HtrA2 и Smac/DIABLO, высвобождающимися во время апоптоза.
И еще о факторах выживания
Межклеточные сигналы регулируют деятельность клеток, в том числе и апоптоз. Необходим контроль, гарантирующий, что отдельные клетки ведут себя во благо всего организма, в противном случае их нужно удалить. Например, сигнальные белки, такие как Fas-лиганд, активируют рецепторы смерти и тем самым инициируют внешний путь апоптоза. Напротив, существуют факторы выживания — внеклеточные сигнальные молекулы, которые ингибируют апоптоз. Некоторые клетки требуют непрерывной сигнализации от других клеток, чтобы выживать. И это, по-видимому, помогает обеспечить жизнь только нужных клеток.
Гибель нервной ткани
Нервные клетки вырабатываются избыточно в развивающейся нервной системе, а затем конкурируют за ограниченное количество факторов выживания. Эти факторы секретируются клетками-мишенями, к которым подходят нейроны. Нервные клетки, получающие достаточно сигналов выживания, живут, в то время как другие, не получающие нужного количества, умирают. Таким образом, число выживших нейронов соответствует количеству клеток-мишеней, с которыми они соединяются.
Жизнь и смерть у нервных клеток.
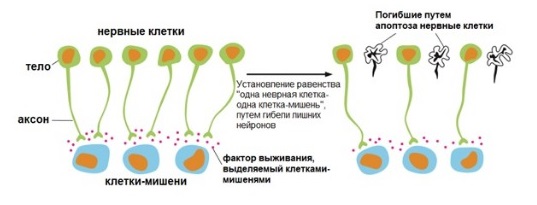
Рисунок 6 | Роль факторов выживания в гибели лишних нервных клеток
Каскады жизни и смерти
Каскад жизни
Факторы выживания для нейронов называются нейротрофическими факторами. Активация рецепторов нейротрофических факторов на пресинаптической мембране аксона приводит к увеличению факторов транскрипции, которые отвечают за образование антиапоптотических белков (Bcl-2, Bcl-xL), супероксиддисмутазы (подавляет повреждение клетки в результате окисления) и белков-ингибиторов апоптоза (IAP).
Каскад смерти
Апоптоз наблюдается в ходе процесса, который называется эксайтотоксичность. Этот процесс происходит при чрезмерной активации глутаматных рецепторов, в результате чего повышается приток Ca2+ в постсинаптические области дендритов. Са2+, попадая в цитоплазму через ионные каналы на плазматической мембране и на эндоплазматическом ретикулуме, индуцирует апоптотический каскад, который активирует проапоптотическое белки Bax, Bad и p53. Эти белки действуют на митохондрии так, что повышается ее проницаемость, а в цитоплазму выделяется цитохром С. Это приводит к апоптозу.
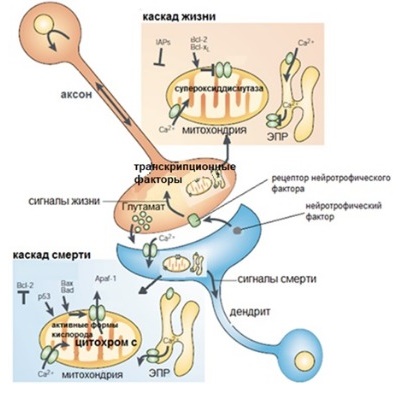
Рисунок 7 | Каскады жизни и смерти
Как убрать апоптотическую клетку?
Апоптоз — очень аккуратный процесс клеточной смерти. Апоптотическая клетка и ее фрагменты не разрываются и не выделяют свое содержимое, а вместо этого остаются нетронутыми. Они съедаются без следов, поэтому воспалительного ответа нет. Апоптотическую клетку поглощают фагоциты. Процесс поглощения зависит от наличия химических изменений на поверхности мембраны клетки.
К таким изменениям относится наличие фосфатидилсерина, который служит сигналом для фагоцитарных клеток. Этот фосфолипид обычно расположен только на внутренней поверхности плазматической мембраны, а при апоптозе переворачивается на наружную поверхность. Макрофаги не фагоцитируют здоровые клетки, хотя у здоровых клеток на их поверхности есть некоторый фосфатидилсерин.
Но здоровые клетки имеют еще и такие сигнальные белки на их поверхности, которые блокируют фагоцитоз. Таким образом, помимо наличия сигналов, таких как фосфатидилсерин, которые стимулируют фагоцитоз, апоптотические клетки должны инактивировать сигналы типа «не ешь меня», блокирующие фагоцитоз.
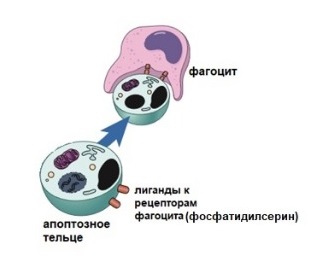
Рисунок 8 | Фагоциты удаляют апоптотическую клетку или ее части
Уклонение от апоптоза
Нарушение механизма клеточной смерти ключевой признак онкологического заболевания. Опухолевые клетки могут использовать различные механизмы для подавления апоптоза и приобретения устойчивости к апоптотическим агентам. Например, может наблюдается повышенная экспрессия антиапоптотических белков (Bcl-2) или мутации в генах проапоптотических белков (Вах).
Дефекты апоптоза могут позволить эпителиальным клеткам выживать во взвешенном состоянии без прикрепления к внеклеточному матриксу, что способствует метастазированию. Они также способствуют устойчивости перед цитолитическими Т-клетками и натуральными киллерами (NK), атакующими опухоли. Эти дефекты играют важную роль в устойчивости к лечению химиотерапией и лучевой терапией, увеличивая порог смерти клеток и требуя более высоких доз агентов, убивающих опухоль.
Успешное удаление раковых клеток с помощью нехирургических средств в конечном итоге достигается путем индукции апоптоза. Все цитотоксические противораковые средства, которые в настоящее время используются в клинических целях, вызывают апоптотическую гибель злокачественных клеток.
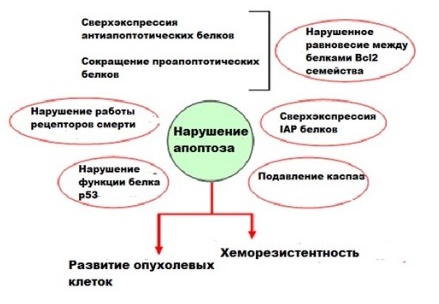
Терапия онкологических заболеваний и апоптоз
Ингибирование апоптоза лежит в основе развития всех опухолей. А значит, наиболее очевидной стратегией лечения является нацеленность на причины, которые подавляют гибель клеток. Для преодоления антиапоптотического эффекта белков Bcl-2 и Bcl-xL в опухолевых заболеваниях существует три стратегии:
- прекращение транскрипции их генов,
- разрушение мРНК с помощью антисмысловых олигонуклеотидов,
- атака мелкомолекулярными препаратами.
Стратегия первая
Некоторые стероиды и ретиноиды активируют транскрипционные факторы, которые регулируют синтез мРНК. Они представляют собой потенциально «лекарственные» модуляторы транскрипции гена Bcl-2 и Bcl-xL. Например, экспрессия Bcl-2 зависит от эстрогена в молочной железе.
Следовательно, антиэстрогены, такие как тамоксифен, ингибируют экспрессию Bcl-2 в клеточных линиях рака молочной железы, способствуя развитию чувствительности к цитотоксическим противоопухолевым препаратам, таким как доксорубицин.
Стратегия вторая
Антисмысловые олигонуклеотиды — цель на мРНК. Антисмысловые олигонуклеотиды (Antisense oligonucleotides) представляют собой короткие последовательности одноцепочечной ДНК, которые могут связываться с мРНК, что сопровождается ее разрушением.
Один из перспективных препаратов - облимерсен натрия. Он представляет собой натриевую соль фосфоротиоатного антисмыслового олигонуклеотида. Препарат ингибирует мРНК гена Bcl-2. Он был успешно протестирован в сочетании с другими противораковыми агентами при различных типах рака, таких как множественная миелома, мелкоклеточный рак легких, меланома и неходжкинская лимфома.
Модификация искусственных нуклеотидов делает нуклеотидную цепь устойчивой к расщеплению нуклеазами и повышают период полувыведения в организме. В данном случае использовалось добавление фосфоротиоата (PS) в основную цепь (замена одного из кислородных остатков фосфатной цепи на серу)
Стратегия третья
BH3 миметики — это вещества, связывающиеся с рецепторами белков, на которые действуют сами BH3. Они необходимы для активации апоптоза. Эти белки нейтрализуют антиапоптотическое белки Bcl2 или активируют Bak и Bax. Разработаны BH3-имитирующие молекулы, выполняющие те же функции, что и BH3 белки.
Например, вещество ABT-737, которое ингибирует Bcl2-белки. BH3-белки, взаимодействуют с длинной гидрофобной канавкой в белке Bcl2, тем самым инактивируя последние. Препарат ABT-737 был спроектирован с помощью кристаллической структуры этой канавки.
Р53 в качестве лекарственной цели
Ген белка р53 отключен из-за мутаций примерно у 50% всех злокачественных опухолей. Повышение производства белка р53 в клетке может быть методом противораковой терапии. В нормальных клетках белок p53 обычно поддерживается на низком уровне, поскольку он подвержен ингибированию белком MDM2. В ответ на повреждения ДНК p53 видоизменяется.
Это позволяет ему избежать контроля MDM2 и начать накапливаться в клетке. Содержание гена MDM2 увеличено в некоторых типах опухолей, поэтому активного белка р53 становится меньше. Взаимодействие между p53 и MDM2 является мишенью в противораковой терапии. С этой целью были разработаны препараты, которые блокируют белок-белковые взаимодействия. Среди них Nutlin-3, ингибирующий взаимодействие p53/MDM2.
Большая часть того, что известно об апоптозе, стала понятна только недавно. Основная идея разработки терапевтических препаратов для лечения рака основана на том факте, что поврежденные клетки обычно встают на путь апоптоза, поддерживая нормальное для ткани количество клеток. Однако это явление сильно нарушается в раковых клетках. Обнаружение ключевых участников апоптоза и их взаимодействие с другими значимыми участниками создает условия для поиска новых методов терапии рака.
Источник: medach.pro